- Record: found
- Abstract: found
- Article: found
Definition and Management of Segmental Pulmonary Hypertension
brief-report
Konstantinos Dimopoulos , MD, MSc, PhD, FESC
1
,
,
Gerhard‐Paul Diller , MD, PhD, FESC
2 ,
Alexander R. Opotowsky , MD, MPH, MMSc
3 ,
Michele D'Alto , MD, PhD, FESC
4 ,
Hong Gu , MD, PhD
5 ,
George Giannakoulas , MD, PhD
6 ,
Werner Budts , MD, PhD, FESC
7
,
8 ,
Craig S. Broberg , MD
9 ,
Gruschen Veldtman , FRCP, MBChB
10 ,
Lorna Swan , MBChB, FRCP, MD
1 ,
Maurice Beghetti , MD, FESC
11 ,
Michael A. Gatzoulis , MA, MD, PhD, FESC, FACC
12
04 July 2018
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Introduction
The World Pulmonary Hypertension Symposium in 2013 (Nice, France) introduced a new
entity in the classification for pediatric and adult patients called “segmental pulmonary
hypertension (PH).”1 Segmental PH was described in the international 2015 guidelines
as PH “observed in discrete lung areas perfused by aortopulmonary collaterals in congenital
heart diseases such as pulmonary or tricuspid atresia,”2 while the proceedings of
the Nice World Symposium1 defined this as “PH in one or more lobes of one or both
lungs.” Others have defined segmental PH more broadly as PH that does not follow a
homogeneous distribution, with some parts of the pulmonary vasculature being exposed
to higher pressures than others.3
This entity was included under the umbrella of World Heart Organization group 5 (PH
caused by unclear or multifactorial mechanisms), because little is known about its
pathophysiology and response to pulmonary arterial hypertension (PAH) therapies.1,
2 Segmental PH is most commonly encountered in patients with congenital heart disease
(CHD) and carries notable similarities to PAH (Group 1.4.4, PAH associated with CHD)
and group 4 of the PH classification (Group 4.2.4 PH in patients with congenital pulmonary
artery [PA] stenoses), yet there is no systematic description of the broad spectrum
of conditions encompassed by this entity or its distinct pathophysiological features
and how these may affect management.
We present herewith a consensus statement on segmental PH, including a working definition,
range of conditions that may be classified under this entity, description of pathophysiology
in terms of pulmonary vasculature, cardiovascular anatomy, and management principles.
Definition and Classification of Segmental PH
The term “segmental” indicates that segments of the lung rather than the entire lung
are affected by pulmonary vascular remodeling and PH (ie, parts of 1 or more lobes,
a single lung, etc.). As such, at least 1 segment of the pulmonary vasculature is
nonhypertensive. While macroscopic branch pulmonary artery stenosis may be associated
with the development of segmental PH, the term “segmental PH” does not refer to disease
of the segmental pulmonary arteries (ie, stenosis of medium‐sized pulmonary arteries
that run alongside segmental bronchi) per se, though segmental PH is often observed
in that setting (eg, Alagille syndrome). A unified definition of segmental PH should
include the following concepts:
1
A condition of 1 or more but not all segments of the lung.
2
Each hypertensive area of the lung may present with PH of different severity.
3
Symptoms of segmental PH often depend on the severity of ventilation‐to‐perfusion
(V/Q) mismatch and right (subpulmonary) heart failure.
In patients with segmental PH, significant differences in lung perfusion in various
lung segments are likely to adversely affect V/Q ratio and blood oxygenation. This
is distinct from inhomogeneity in the V/Q observed in normal people (eg, greater towards
the apex).
Lung segments supplied by collaterals arising from the aorta receive partially (or
fully) oxygenated blood, hence, again causing an increase in physiological dead space.
Co‐existing right‐to‐left shunting is common and contributes further to V/Q mismatch.
4
Affected lung segments may or may not be in direct communication with the right (or
subpulmonary) ventricle.
5
Segments of the pulmonary vasculature that are in direct (unobstructed) communication
will have identical pulmonary pressures, even if peripheral resistances differ. This
is also true in the case of pulmonary vein stenoses, which can raise pulmonary wedge
pressure in 1 or often more lung segments, but will not cause a segmental rise in
pulmonary arterial pressure or a rise in PVR.
6
Separate detailed assessment of PA pressures and calculation of pulmonary vascular
resistance (PVR) in each lung segment is required to fully understand lung pathophysiology
and the severity of PH in these patients. This often, however, proves difficult even
in expert hands.
In this article, we propose the following definition of segmental PH: Segmental pulmonary
hypertension encompasses any condition with abnormal underlying cardiac or vascular
anatomy, usually including varied sources of pulmonary blood supply, which results
in distal pulmonary vascular disease that affects various lung segments to differing
degrees.
Lung perfusion and the relation between the heart and the pulmonary vascular tree
varies significantly within the spectrum of segmental pulmonary hypertension and may
affect presentation and the response to therapies. Hence, we propose the following
classification for segmental PH:
Right ventricle (RV) communicating directly with the entire pulmonary vascular bed
(eg, large ventricular septal defect [VSD] with peripheral pulmonary stenosis (PS)
and ventriculo‐arterial concordance).
RV supplies part of the pulmonary vascular bed (eg, congenital absence/interruption
of a pulmonary artery supplied by large collaterals/isolated pulmonary artery of ductal
origin or a patent ductus arteriosus [PDA], hemitruncus arteriosus).
RV with no direct communication with the pulmonary vascular bed:
With well‐formed (native) PAs (eg, truncus arteriosus with PA stenosis);
With ill‐formed PAs and a pulmonary circulation supplied by collateral arteries, a
PDA, or surgical shunts (eg, tetralogy of Fallot [TOF] with pulmonary atresia, often
referred to as complex pulmonary atresia throughout this article).
There are cases in which the subpulmonary ventricle is a morphologic left ventricle;
the current article uses the term RV to refer to the subpulmonary ventricle, independent
of anatomic characteristics.
Conditions in Which Segmental PH May Develop
Congenital lesions that may lead to segmental PH include complex pulmonary atresia,
hemitruncus arteriosus, absence/atresia of a single pulmonary artery, and an anomalous
pulmonary artery from the aorta feeding a single lung segment (Figure 1). Moreover,
any large post‐tricuspid cardiac defect (eg, VSD atrioventricular septal defect, PDA,
aortopulmonary window, or truncus arteriosus) that may lead to increased PVR (ie,
Eisenmenger physiology) can result in segmental PH when peripheral PA stenosis is
present, whether naturally or because of failed PA banding. In these cases, the PA
stenosis effectively “protects” some but not all segments of the lung from developing
pulmonary vascular disease. Finally, surgical shunts (eg, Potts or Waterston shunt)
that may supply only part of the pulmonary vascular bed (eg, disconnected PAs), or
cause localized branch PA stenosis, can lead to segmental PH. Specific examples of
these congenital variants are considered below. Of note, in conditions such as Alagille
syndrome, branch PA stenosis may result in PH in unobstructed segments of the lung
in the absence of an intracardiac defect, somewhat resembling chronic thromboembolic
PH in pathophysiology. Such conditions are mentioned in the international PH classification
under group 4 (chronic thromboembolic PH and other PA obstructions, group 4.2.4).2
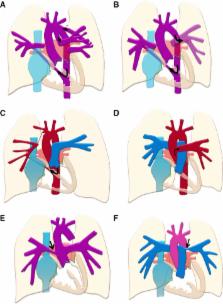
Figure 1
Schematic examples of segmental pulmonary hypertension. In (A), complex pulmonary
atresia, with nonconfluent pulmonary arteries supplied by a patent ductus arteriosus
and major aortopulmonary collateral arteries (MAPCAs) from the descending aorta (arrows),
is shown. In (B), the left pulmonary artery is supplied by a Potts shunt and the right
by a MAPCA from the descending aorta (arrows). Potts (descending aorta to left pulmonary
artery) and Waterston (ascending aorta to right pulmonary artery) shunts are difficult
to size and are therefore more likely to cause pressure and volume overload of the
lung segments supplied, over time leading to the development of segmental pulmonary
hypertension. In (C), unilateral absence of right pulmonary artery or isolated right
pulmonary artery of ductal origin. There is a small outpouching of the innominate
artery and a ductal remnant to the isolated pulmonary artery. The latter is supplied
by a large MAPCA from the descending aorta (arrow), which may cause pulmonary hypertension
(PH) to develop in the right lung. Pulmonary hypertension may also develop in the
left lung only. In (D), hemitruncus arteriosus, with the left pulmonary artery arising
from the hemitruncus (arrow) and the right from the main pulmonary artery in direct
communication with the right ventricle. Only the left lung is hypertensive. In (E),
common arterial trunk with stenosis of the origin of the right pulmonary artery (arrow)
is shown. In this case, only the left lung is hypertensive. In (F), there is a large
ventricular septal defect (VSD), and stenosis of the origin of the left pulmonary
artery (arrow). In this condition, shunting through the nonrestrictive VSD is likely
to cause distal PH and pulmonary vascular disease in the right, but not the left lung.
Right‐to‐left shunting may be caused by right heart remodeling as a result of the
combination of proximal left branch PA stenosis and distal right pulmonary vascular
disease (PH).
Pulmonary Atresia
Pulmonary atresia is encountered in association with numerous congenital heart defects,
and there is no universal agreement on its anatomic and clinical classification.4,
5 Two main forms exist. The first, pulmonary atresia with a VSD and a biventricular
circulation, is generally considered part of the spectrum of TOF (Figure 2). Pulmonary
atresia may alternatively present with an intact ventricular septum, or in the setting
of more complex anatomy (eg, transposition of great arteries, tricuspid atresia, etc).
Under the umbrella of pulmonary atresia, there is significant variability in the anatomy
of the PAs. When branch PAs are of adequate size and confluent, it allows for an anatomic
repair by implantation of a RV to PA conduit.6 Alternatively, when the branch PAs
are hypoplastic, construction of a Blalock‐Taussig or a central shunt may be needed
to promote growth of the PAs to eventually allow conduit repair, with or without unifocalization.
Figure 2
Tetralogy of Fallot with complex pulmonary atresia and previous bilateral Blalock
Taussig shunts (A, arrows). The right pulmonary artery (RPA) is hypertensive and severely
dilated, while the left pulmonary artery (LPA) is supplied by a relatively small collateral
and is of normal caliber. This patient has segmental pulmonary hypertension, with
pulmonary hypertension of various severities in different segments of the lung (eg,
right mid and lower), while other segments are normotensive and may be hypoperfused.
In (B), chest radiograph of the same patient shows a boot‐shaped heart with inhomogeneous
pulmonary vascular markings.
In TOF with “complex” pulmonary atresia, the PAs are typically hypoplastic and have
reduced bronchopulmonary segments; they may be nonconfluent and supplied by a PDA,
or represent major systemic‐to‐pulmonary collateral arteries anatomically distinct
from the PDA (also termed major aortopulmonary collateral arteries [MAPCAs] Figure 3).6
In extreme cases, the entire intrapericardial pulmonary arterial tree is absent and
blood is supplied to the lungs by collateral arteries only. Some patients may be amenable
to surgical repair.7, 8, 9 “Unifocalization” is a staged approach aimed at reconstructing
the PAs and perfusing them by means of modified Blalock‐Taussig shunts with the ipsilateral
subclavian arteries, or with other shunts as dictated by specific PA anatomy. Thereafter,
the reconstructed PAs can be connected to the RV with a RV‐PA conduit, with subsequent
closure of the VSD, as long as proximal PA pressure is low enough.
Figure 3
Computed tomography scan of a patient with tetralogy of Fallot and complex pulmonary
atresia. In (A), major aortopulmonary collateral arteries (MAPCAs, arrow) arising
from the descending aorta (DAo), some of which are large and may cause pulmonary hypertension
in the areas supplied, while others are narrow and have required stenting to ensure
adequate perfusion in the respective lung segments. In (B), a large MAPCA (arrow)
arising from the ascending aorta (AAo) and supplying a hypertensive lung segment is
seen. Numerous other MAPCAs are seen within the mediastinum. Ascending aortic dilation
is apparent.
Patients with pulmonary atresia who do not undergo early operation or have only received
a palliative intervention or partial unifocalization develop segmental PH as a result
of large MAPCAs or palliative shunts (especially Waterston or Potts anastomoses, which
are challenging to size) causing excessive flow and shear stress to certain, but not
all, lung segments. Patients in whom unifocalization has been possible, but PA pressures
are high, may receive an RV‐PA conduit but the VSD is kept open (or closed using a
valved patch) as a potential “relief valve” for the RV. Finally, even in patients
with “successful” repair, residual or recurrent peripheral PA stenoses are not uncommon
after unifocalization, and pulmonary vascular disease in segments previously supplied
by large MAPCAs may result in segmental PH.
Unilateral Absence of PA or Isolated PA of Ductal Origin
Unilateral “absence” of a PA is very rare. The term “absent” PA is not accurate, as
a hilar PA is typically present and supplied by a PDA or large collaterals but not
the main PA, often leading to the development of PH in a single lung.10, 11 Several
authors have used the terms “isolated PA of ductal origin” or “unilateral ductal origin
of a PA” to more accurately describe this condition. Strategies to rehabilitate the
isolated PA have been reported.12, 13, 14, 15, 16, 17
Its prevalence as an isolated lesion is estimated at 1 in 200 000‐to‐300 000 adults,10,
18, 19, 20 and 80% of reported cases involving the left PA have been associated with
coexisting CHD, such as TOF or truncus arteriosus.10, 19 In 2011, a review of the
literature reported 352 cases of unilateral “absence” of pulmonary artery; two thirds
(n=237) were associated with other CHD.21 PH is present in 44% of cases and, in conjunction
with the underlying CHD, affects appropriate management and outcomes for these patients.18,
19, 22 PH may occur as a result of increased flow to the “healthy” lung, or in the
“disconnected” lung supplied by large collaterals or a large PDA (Figure 4).22, 23,
24
Figure 4
Anomalous origin of the left lower pulmonary artery (black arrow) from the descending
aorta (DAo). The distal branches of the left lower pulmonary artery (white arrow)
are dilated. There is segmental pulmonary hypertension limited in the left lower lung
lobe.
Hemitruncus Arteriosus
Hemitruncus arteriosus refers to the abnormal origin of a single PA from the ascending
aorta, with normal origin of the contralateral PA from the main PA; the latter is
normally connected to the RV (Figure 5).5, 6 Separate ventriculo‐arterial junctions
and separate aortic and pulmonary arterial valves mean that hemitruncus is a different
entity from common arterial trunk (truncus arteriosus). The lung supplied by a normal‐sized,
unobstructed PA originating from the aorta is typically pressure and volume overloaded
early in life and pulmonary vascular disease develops. Mortality is high in the first
year of life (up to 70%) without timely repair.7 A small proportion of unrepaired
patients do survive to adulthood and beyond, with segmental (eg, unilateral) PH.
Figure 5
Hemitruncus arteriosus. In (A), the left pulmonary artery (LPA) is seen on cardiac
magnetic resonance arising from the ascending aorta (AO). In (B), the right pulmonary
artery (RPA) arises from the main pulmonary artery (MPA). In this situation, the left
lung is hypertensive while the right is not, in the absence of associated CHD (eg,
large VSD or PDA to the RPA). CHD indicates congenital heart disease; PDA, patent
ductus arteriosus; VSD, ventricular septal defect.
Truncus Arteriosus With Stenosis or Hypoplasia of a Single PA or Branches
Truncus arteriosus is characterized by a common arterial trunk, giving rise to both
the systemic and pulmonary circulation. The PAs originate from the arterial trunk
in various patterns25, 26 and the presence of normal‐sized PAs results in bilateral
PH, unless there is stenosis of the origin of 1 or both PAs (present in half of the
patients). When branch pulmonary artery stenosis is severe, it may “protect” the respective
lung from developing pulmonary vascular disease; segmental PH develops in the contralateral
lung.26, 27 Historically, patients without PA stenosis would often undergo banding
of the PAs before definitive repair, while at the current time early, complete repair
is the treatment of choice. A “slipped” PA band is not uncommon and has the same effect
as congenital branch PA stenosis, protecting 1 but not both lungs. This may result
in segmental PH.
Large Post‐Tricuspid Defects With Peripheral Pulmonary Stenosis
Patients with large post‐tricuspid shunts, such as a VSD, atrioventricular septal
defect, PDA, or aortopulmonary window, are expected to develop pulmonary vascular
disease in the absence of timely repair or effective PA banding. In unrepaired patients,
or patients with a large residual post‐tricuspid shunt, the presence of significant
peripheral PA stenoses may “protect” part of the lung from the abnormal shear stress,
resulting in segmental PH (Figure 6).
Figure 6
Tetralogy of Fallot and complex pulmonary atresia after conduit repair and closure
of the ventricular septal defect, but with a significant residual shunt. In (A), there
is significant pulmonary hypertension in the left lung with a dilated left pulmonary
artery (LPA), while the right lung is “protected” by a stenosed right pulmonary artery
(RPA). In (B), severe cardiomegaly with prominent pulmonary vascular marking in the
hypertensive left lung are seen, but not in the right lung. Kyphoscoliosis, common
in this group of patients, is also apparent and can independently impair pulmonary
gas exchange and predispose towards pulmonary vascular remodeling in severe cases.
The remainder of this article will focus on the most common cause of segmental PH:
TOF with complex pulmonary atresia.
Pathophysiology
Histology of the Pulmonary Circulation
The pathophysiology of segmental PH has not been well studied, but is likely to be
the result of increased shear stress from excessive flow and pressure by large collaterals
or abnormal origin of the PAs (similar to what is observed in patients with a large
ventricular septal defect, patent ductus arteriosus, and common arterial trunk).28
Moreover, hypoplasia of the pulmonary vascular bed is described in children. Thiene
et al demonstrated that lung segments perfused by large, nonstenotic systemic collateral
arteries in patients with pulmonary atresia had features of proliferative pulmonary
vascular disease, including medial hypertrophy, intimal proliferation, and even plexiform
lesions.29 Different degrees of pulmonary vascular disease were detected in various
lung segments of a patient with multifocal PA supply. In the oldest child, who died
at age 10 years, “obliterative” pulmonary vascular disease had developed. Haworth
and Reid studied the lungs of neonates and children with pulmonary atresia, both those
with intact ventricular septum and those with a VSD; they reported impaired lung development,
with few and small pulmonary arteries with abnormally thin muscle coat.30 Hence, the
rise in segmental PVR in these patients may be the combined effect of pulmonary vascular
disease and abnormal development of the pulmonary circulation, at both macroscopic
and microscopic level. Understanding the pathophysiology of segmental PH is important
for designing new treatment strategies for these patients.
Effects of Segmental PH on Pulmonary Physiology, Oxygen Tissue Delivery, and the Heart
In segmental PH, different areas of the lung receive different amounts of blood flow,
at different pressures, and from different sources. Asymmetric perfusion results in
ventilation/perfusion (V/Q) imbalance, which has been associated with adverse outcome
in children with pulmonary atresia.31 V/Q mismatch results in a reduction in peripheral
oxygen delivery at equivalent hemoglobin concentration and inefficient ventilation,
imposing an additional workload on the heart and lungs. The coexistence of an intracardiac
right‐to‐left shunt, with desaturated blood “bypassing” pulmonary gas exchange in
the lung, causes further physiological dead space and V/Q mismatch, as does perfusion
of the lung by partially or fully saturated blood from the aorta, with little or no
gas exchange occurring in such areas. Finally, gas exchange in patients with complex
pulmonary atresia is also affected by the presence of hypertensive segments of the
lung that are often adjacent to hypoperfused segments (supplied by small or stenosed
collaterals).
There are, therefore, substantial differences in cardiac physiology in various types
of segmental PH compared with other types of PH. The proposed classification of segmental
PH reflects this and, specifically, the relation of the RV to the pulmonary circulation.
In patients with complex pulmonary atresia or truncus arteriosus, the RV does not
eject blood into the pulmonary circulation, and hence it is not directly affected
by changes in pulmonary pressure and resistances. However, RV hypertrophy, dilatation,
and dysfunction and tricuspid regurgitation can result from the large ventricular
communication and resultant systemic RV pressures, ultimately leading to right heart
failure independent of whether PH per se (ie, proximal PA pressure >25 mm Hg) is present.
Moreover, systemic hypoxemia and chronic cyanosis affect all major organs, including
the myocardium, producing an adverse effect on both the right and left ventricles.
Indeed, the aorta receives a large quantity of blood, equal to the sum of the systemic
and pulmonary blood flow. As patients age, both systemic ventricular and aortic dilation
are common; aortic regurgitation can develop as a result of changes in aortic geometry.
Some have expressed concern that pulmonary vasodilator medications (targeted PAH therapies)
could further volume load the heart by significantly augmenting pulmonary blood flow.
This has not been substantiated by the limited data available, with few patients followed
for relatively short periods of time.3 Experience suggests, however, that life expectancy
of patients with progressive symptoms related to complex pulmonary atresia may make
long‐term consequences of such therapies on cardiac structure and function less relevant
to clinical practice.
For patients in whom the RV is directly connected to the entire pulmonary circulation,
the effect of segmental PH on the RV is in addition to the load imposed by obstructive
lesions (peripheral pulmonary stenosis, RV‐PA conduits) themselves, the effects of
previous cyanosis, surgical injury, and the burden of residual lesions (eg, a VSD).
The interaction between the pulmonary circulation and the heart becomes even more
complicated when the RV is “connected” to part rather than all of the pulmonary vascular
tree; this merits further study, though disease rarity and heterogeneity present obstacles
to systematic investigation. Finally, hemoptysis is not uncommon in pulmonary atresia,
especially in patients with large hypertensive PAs. Understanding the anatomy of the
PAs and collateral vessels is essential when considering embolization.
Assessment of Segmental PH in TOF With Pulmonary Atresia
Patients with complex pulmonary atresia in TOF are cyanosed at birth, because of obligatory
mixing of oxygenated and deoxygenated blood within the aorta.6 Low or decreasing systemic
oxygen saturations in complex pulmonary atresia may be related to PH or inadequate
lung perfusion (eg, stenosis of collateral arteries or of a previously placed shunt).
The first heart sound is normal but the second heart sound is typically single and
loud, not because of PH, but because of the anterior positioning of the enlarged aorta
below the chest wall. Continuous murmurs relating to the PDA or MAPCAs can be heard
throughout the chest but become systolic and less prominent when PVR increases in
the vascular bed distal to such vessels. Aortic regurgitation and stenosis may also
develop in older patients, because of progressive aortic dilation.
The ECG usually shows signs of RV hypertrophy and right atrial dilatation. Echocardiography
is essential when screening for PH, but is not sufficient for firmly establishing
the diagnosis of segmental PH, and clearly less adequate for full definition of branch
PA anatomy. It is important to remember that, in the absence of communication between
the RV and PAs, neither estimates of RV pressure (using the tricuspid regurgitation
velocity by Doppler) nor any property of the RV (eg, size or function) provide any
insight into the state of the pulmonary circulation. However, Doppler can be used
to interrogate flow through collateral vessels or surgical shunts, providing rough
estimates of gradients between the aorta and the PAs. Continuous flow with a high
peak velocity (>4 m/s in the presence of normal systemic pressures) can be taken as
an indirect sign that no significant rise in pressure has occurred in the segment
of lung fed by the interrogated shunt; a low peak velocity, in contrast, with a shunt
that is mainly systolic suggests that PH and pulmonary vascular remodeling are likely.
Other noninvasive investigations can provide valuable information in the assessment
of segmental PH in complex pulmonary atresia. The chest radiograph may show a “boot‐shaped”
heart, a combination of absent central PAs with an elevated apex because of RV hypertrophy,
sometimes made more prominent by a right‐sided aortic arch. The chest radiograph also
provides information on the relative size of the central pulmonary arteries, which
are often dilated when hypertensive, as well as the relative perfusion of various
segments of the lung parenchyma (patchy and inhomogeneous pulmonary vascular markings
with hypoperfused darker areas versus well‐perfused brighter areas). Chest computed
tomography pulmonary angiography provides more detailed information based on the same
principles, and is a test of choice for obtaining anatomic information on the aorta,
PAs, and collateral arteries/shunts with excellent spatial resolution. In adult patients,
a large MAPCA or PDA without stenosis invariably implies that the corresponding lung
segment is hypertensive. Moreover, PA dilatation (with or without in situ thrombosis)
is suggestive of PH. Cardiac magnetic resonance provides valuable information on cardiovascular
anatomy, including the morphology and size of central PAs, the presence and function
of large collateral arteries and shunts, as well ventricular function, aortic dimensions,
and the function of the aortic valve.32
Cardiac catheterization remains the criterion standard for assessing segmental PH
but can be a long and laborious process, requiring administration of potentially large
amounts of contrast medium and is best interpreted in conjunction with the noninvasive
imaging data. A full hemodynamic study in patients with complex pulmonary atresia
requires pump contrast injections in the aorta for the identification of MAPCAs and
other shunts, followed by selective angiography and hemodynamic assessment of each
vessel. Collateral vessels need to be adequately intubated to assess pressures in
the respective distal pulmonary segments and the pressure gradients across the collaterals;
some operators use coronary pressure wires for this purpose. Damage to important collateral
vessels and shunts can have devastating consequences and cardiac catheterization should
be undertaken in expert centers, only when essential and as a prelude to surgery or
other intervention being considered for patients with symptomatic decline.
The anatomy of the pulmonary vascular tree influences the development of PH, response
to therapy, and long‐term outcome and is key in deciding when a patient can undergo
biventricular repair. Patients with well‐developed central pulmonary arteries provided
by a ductus arteriosus have a significantly better prognosis and are more often amenable
to primary repair with closure of the VSD and reconstruction of the right ventricular
outflow tract by patch or conduit, compared with patients who are dependent on MAPCAs.
Patients with more hypoplastic central pulmonary arteries may require palliation (eg,
with a modified Blalock Taussig shunt) before proceeding to repair. In patients with
MAPCAs, the number, size, and distribution of collateral arteries can vary considerably
and influence the development of PH. Hence, it is important to perform imaging of
the vascular bed early in life to assess how many of the segments are perfused centrally,
and carefully assess patients for biventricular repair. Moreover, there is a relation
between the vascular bed available and PH, and extensive, unrestrictive collateral
vessels, which can lead to congestive heart failure early in life, can eventually
result in PH.
The interplay between flow and pressure in different lung segments makes calculation
of PVR a key to a deep understanding of underlying pathophysiology. However, in the
presence of multifocal blood supply, calculation of PVR may be challenging (or impossible)
and magnetic resonance imaging or quantitative nuclear perfusion imaging may be of
help in assessing pulmonary blood flow. In the setting of a strictly unifocal pulmonary
blood supply (eg, single large PDA or MAPCA with confluent PAs), central PAs should
be entered with the catheter to estimate PVR. As previously stated, prior advanced
imaging with cardiac magnetic resonance imaging or computed tomography can delineate
the target vessels and guide the invasive cardiac catheterization.
Clinical Management of Segmental PH: Surgical, Interventional, and Medical Therapy
In children born with complex pulmonary atresia with a VSD, the aim is to create an
adequate pulmonary artery tree supplied by the right ventricle and close the VSD.
Biventricular repair can be achieved through unifocalization (when PAs are not confluent)
and implantation of a RV‐PA conduit, while in a minority of cases a Fontan‐type palliation
or other palliative procedures may be undertaken.33, 34, 35 In unrepaired adults,
unifocalization is often not feasible, and the presence of segmental PH precludes
complete repair. A challenge is to ensure the lung segments are neither hypoperfused
(excessive stenosis) nor hyperperfused (unrestrictive flow via large collaterals).
Palliative shunts or dilatation +/− stenting of existing but stenosed MAPCAs or surgical
shunts can be undertaken in symptomatic patients who present with hypoperfused lung
segments.31, 36, 37 Experience is paramount, given the risks of immediate or delayed
“reperfusion” pulmonary edema, hemoptysis, or other potentially catastrophic outcomes
of injudicious interventions.
There are few reports on the effects of PAH medications (advanced therapies) in segmental
PH. A multicenter study by Schuuring et al on 7 patients with segmental PH caused
by pulmonary atresia reported a significant improvement in functional class and exercise
tolerance (6‐minute walk test distance improved by 62 m) with the endothelin‐receptor
antagonist bosentan.3 Another observational study by Lim et al on 5 adult patients
with complex pulmonary atresia or severe pulmonary stenosis and MAPCAs treated with
the phosphodiesterase‐type 5 inhibitor sildenafil reported this therapy to be well
tolerated in 4 of 5 patients, with a good clinical response to treatment in those
patients.38 Yamamura et al presented 2 children with segmental PH after pulmonary
atresia treated with bosentan, with an improvement in symptoms, hemodynamics, and
brain natriuretic peptide concentration.39 Yasuhara and Yamagishi presented 3 patients
who were likely to have segmental PH.40 A pediatric patient developed PH after repair
of pulmonary atresia (unifocalization). PA pressure improved through percutaneous
treatment of peripheral pulmonary stenosis and combination medical PAH therapy. An
adult with unrepaired TOF, severe pulmonary stenosis, hypoplastic PAs, and MAPCAs
had raised (peripheral) PA pressures and was treated with an endothelin‐receptor antagonist,
with alleviation of symptoms of exercise intolerance and improvement in quality of
life. A third case of an adult with TOF and a hypoplastic left PA repaired at the
age of 5 years presented at 27 years with an occluded left PA and raised PA pressures
on the right PA. Treatment with an endothelin‐receptor antagonist led to worsening
hypoxemia, which the authors attributed to exacerbated V/Q mismatch or volume overload.
Apostolopoulou et al reported the use of PAH therapies in 3 adult patients with unrepaired
TOF and pulmonary atresia. PAH treatment resulted in an improvement in functional
class and no adverse effects.41
Thus, the effect of PAH therapy in patients with segmental PH remains a matter of
debate. While some evidence suggests this approach may be promising, there have been
cases where therapies were not tolerated.38, 39, 42 An increase in pulmonary blood
flow may, theoretically, overload the left ventricle and this should be taken into
account when considering PAH therapies in patients with established left ventricular
dysfunction and/or aortic stenosis or regurgitation. Limitations in the assessment
of PVR and the effect of peripheral pulmonary stenoses also present a challenge in
identifying patients who may benefit the most from PAH therapies. The use of PAH‐specific
therapies has been reported in a few cases of unilateral absence of a pulmonary artery;
however, the evidence is not strong enough to allow for firm recommendations.
To Which World Health Organization PH Group Does Segmental Pulmonary Hypertension
Best Belong?
Segmental PH appears to share histological features with PAH (group 1). Yet it also
overlaps with PH in patients with pulmonary arterial obstructions, including congenital
branch pulmonary artery stenosis or chronic thromboembolic PH (group 4), in terms
of both the heterogeneity of histologic changes and perfusion, along with the potential
for structural intervention in a subset. Some types of group 5 PH, such as fibrosing
mediastinitis or pulmonary arterial compression by tumors, may also present with segmental
PH. Intimal proliferation has been described in hypertensive lung segments of young
patients with pulmonary atresia, although most of the available data are in infants.
While medial hypertrophy and intimal proliferation in hypertensive lung areas in pulmonary
atresia resemble the changes seen in PAH (group 1), the conditions grouped under PAH
share not only histological, but also clinical features and a similar response to
therapy. Hence, at present, it may seem appropriate to classify segmental PH within
group 5, multifactorial PH, reinforcing the complexity and unique physiology of this
condition, and its distinct anatomical and clinical features compared with chronic
thromboembolic PH. However, inclusion within group 1 (PAH related to CHD) may have
the merit of reminding physicians this is a CHD‐related condition, with significant
similarities in pathophysiology to other CHD‐related PAH (eg, chronic cyanosis) and
may help inclusion in future research. Despite notable similarities with other types
of PH, including chronic thromboembolic PH, it is imperative to approach segmental
PH as a separate entity and practice caution when extrapolating information from other
conditions with regard to medical and interventional treatment.
Where Should Patients With Segmental PH Be Managed?
Segmental PH is a complex condition that encompasses a broad spectrum of CHD and,
less commonly, acquired causes. Assessment and interpretation of pathophysiology requires
tertiary expertise both in CHD and PH. Confirmation of segmental PH, assessment of
hemodynamics, and clinical relevance in individual cases and potential benefits of
interventional and/or PAH therapies remain challenging. Patients with segmental PH
should be cared for in tertiary centers with expertise in both PH and CHD patients,
complex noninvasive and invasive investigations, and multidisciplinary support in
terms of imaging, catheter, and cardiac surgical interventions. A broad recommendation
on the use of PAH therapies cannot be made at present given the lack of evidence,
though anecdotal experience suggests these therapies may have a role and may be considered
empirically on an individualized basis in patients with confirmed segmental PH. Because
of significant heterogeneity, coupled with a small patient population, it is unlikely
that adequately powered randomized controlled trials will ever be feasible. However,
well‐structured prospective registries with prespecified baseline and follow‐up protocols
may shed additional light on the natural and unnatural history, and optimal management
of segmental PH.
Author Contributions
Dimopoulos drafted the manuscript. All authors have provided feedback and critically
revised the manuscript and its intellectual content.
Disclosures
Dimopoulos has acted as a consultant and received unrestricted educational or research
grants from Bayer UK, Pfizer, Actelion, and GSK. Diller has received educational/travel
grants from Actelion and Pfizer, and has served on the advisory boards of Actelion,
Germany. Opotowsky has received research grants from Actelion and Roche Diagnostics
and has consulted for Novartis. D'Alto has served on the advisory board of Actelion,
Bayer, United Therapeutics, and GlaxoSmithKline. Gu has received research grants from
Actelion. Giannakoulas has acted as a consultant and/or received unrestricted educational
or research grants from Actelion, Bayer, MSD, GlaxoSmithKline, Lilly, Pfizer, and
United Therapeutics. Budts received a research grant from Actelion. Veldtman received
a single research support from United Therapeutics. Beghetti has served as a consultant
and/or advisory board member for Actelion, Bayer, Eli Lilly, GlaxoSmithKline, Novartis,
and Pfizer and has received investigator‐initiated research funding from Actelion
and Bayer. Gatzoulis has acted as a consultant and received unrestricted educational
or research grants from Bayer UK, Pfizer, and Actelion. The remaining authors have
no disclosures to report.
Related collections
Most cited references39
- Record: found
- Abstract: found
- Article: not found
Isolated unilateral absence of a pulmonary artery: a case report and review of the literature.
Jaap Ottenkamp, Marc A G J Ten Dam, Nico A Blom (2002)
- Record: found
- Abstract: found
- Article: not found
The varied manifestation of pulmonary artery agenesis in adulthood.
K Tsintiris, P Pare, P Panagou … (1995)
- Record: found
- Abstract: found
- Article: not found
Unilateral absence of pulmonary artery: pathophysiology, symptoms, diagnosis and current treatment.
Olga Pechanova, Gabriela Kovacova, Peter Kruzliak … (2013)