- Record: found
- Abstract: found
- Article: found
Diagnosis of Variant Klinefelter Syndrome in a 21-Year-Old Male Who Presented with Sparse Facial Hair
letter
Seda Purnak , M.D.
,
Simin Ada , M.D.,
A. Tülin Güleç , M.D.,
Tugce Bulakbasi Balci , M.D.
1 ,
Feride Iffet Sahin , M.D., Ph.D.
1
25 July 2012
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Dear editor:
Klinefelter syndrome (KS) describes a sex chromosomal aneuploidy caused by the addition
of at least one extra X chromosome to normal male karyotype, XY. It is the most common
disorder of sex chromosomes with a prevalence of one in 600 males1. Variants of KS
are characterized by the addition of an extra X or Y chromosome to classic karyotype
47,XXY. Although somatic malformations and mental retardation are more severe in these
variants, most cases remain undiagnosed till puberty when the symptoms of androgen
deficiency are recognized2,3.
A 21-year-old male presented with sparse facial hair since the onset of puberty. His
medical history revealed cryptorchidism, delayed neuromotor development, and mild
mental retardation recognized in early childhood. His parents were nonconsanguineous,
and there was no history of a genetic disease in the family. On physical examination,
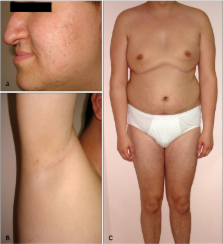
the facial (Fig. 1A) and axillary hair (Fig. 1B) were sparse whereas other body hair
was in normal density. His height and weight were 193 cm and 103 kg, respectively,
and the feminine distribution of the adipose tissue was striking on examination (Fig.
1C). He had dysmorphic features consisting of hypodontia, hypoplastic teeth, prognatism,
short filtrum (Fig. 1A), gynecomastia (Fig. 1C), clinodactily, and fusiform shaped
fingers. Chromosome analysis of a peripheral blood sample disclosed a 48,XXXY karyotype,
compatible with the diagnosis of variant KS.
Detailed hormonal profile analysis revealed a hypergonadotropic hypogonadism, and
mild hypothyroidism. Furthermore, scrotal ultrasonography showed small sized testicles.
Accordingly, the patient was started on testosterone treatment.
Patients with KS may present with a variety of subtle clinical signs of endocrinological,
psychological and orthopedic problems. Thus the diagnosis may be quite challenging
after birth. In infancy, hypospadias, cryptorchidism or small phallus may be helpful
for the diagnosis, while during childhood, speech and language deficits, behavioral
and cognitive problems may serve as diagnostic clues1. After puberty, a relative testosterone
deficiency becomes evident, and suspicion arises when the development of secondary
sexual characteristics is not completed4. The typical male with KS is characterized
by eunuchoid body proportions, abnormally long legs and arm span, feminine distribution
of adipose tissue, absent or sparse facial, body and sexual hair, small testes and
penis. The syndrome may later be diagnosed among adults with azoospermia visiting
infertility clinics2. Correct diagnosis needs careful attention as the syndrome is
mostly overlooked. One study showed that 10% of the cases were recognized prenatally
while 26% were diagnosed in childhood or adult life, leaving 64% of the cases undiagnosed2.
The existence of every extra chromosome in KS worsens the clinical manifestation.
An average to tall stature, ocular hypertelorism, flat nasal bridge, epicanthic folds,
low set ears, prognathism, kyfosis, scoliosis, radioulnar synostosis and clinodactily
are common findings of 48,XXXY variant of KS1,3. Prevalence of this disorder is approximately
1:50,0001,3. The rarity of reports may be due to a phenotype identical to the 47,XXY
phenotype3. Thus, in the literature, the data for 48,XXXY variants is limited, especially
regarding the age of diagnosis and congenital malformations5.
Clinicians should pay more attention to this easily overlooked syndrome in order to
diagnose more boys with KS at an earlier age. The finding of sparse facial and axillary
hair, even though could be familial, is a typical symptom of hypogonadism. Among the
most frequent causes of male hypogonadism, KS should come to mind in the presence
of eunuchoid body proportions, cognitive or dysmorphic facial features. Unfortunately,
our case was left undiagnosed till he visited our clinic with the complaint of sparse
facial hair at the age of 21. When his dysmorphic features came to notice during the
dermatologic examination, a chromosome analysis was performed and the patient was
properly diagnosed as variant KS.
In conclusion, KS and its variants can be recognized by even very indefinite findings
such as sparse facial hair when combined with a careful detailed physical examination.
Thus, we would like to emphasize the importance of a thorough physical examination
during dermatological evaluation of a patient that can disclose any underlying systemic
illness.
Related collections
Most cited references4
- Record: found
- Abstract: found
- Article: not found
48,XXYY, 48,XXXY and 49,XXXXY syndromes: not just variants of Klinefelter syndrome.
Philip Zeitler, Cheryl D'Epagnier, Natalie Ayari … (2011)
- Record: found
- Abstract: found
- Article: not found
Sex chromosome tetrasomy and pentasomy.
M Linden, B Bender, A. Robinson (1995)
- Record: found
- Abstract: found
- Article: not found
Pituitary-gonadal function in Klinefelter syndrome before and during puberty.
J A Salbenblatt, B Bender, M H Puck … (1985)