- Record: found
- Abstract: found
- Article: found
Mitophagy-mediated adipose inflammation contributes to type 2 diabetes with hepatic insulin resistance

Read this article at
Abstract
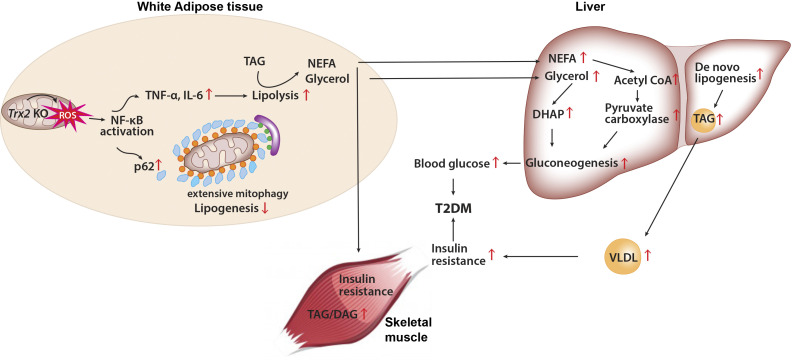
Adipose-specific deletion of mitochondrial redox Trx2 induces excessive mitophagy in white adipose tissue with increased inflammation and increased lipolysis, promoting hepatic glucose production and development of T2DM with hepatic steatosis. Administration of NF-κB inhibitor prevents adipose mitophagy and ameliorates T2DM progression.
Abstract
White adipose tissues (WAT) play crucial roles in maintaining whole-body energy homeostasis, and their dysfunction can contribute to hepatic insulin resistance and type 2 diabetes mellitus (T2DM). However, the mechanisms underlying these alterations remain unknown. By analyzing the transcriptome landscape in human adipocytes based on available RNA-seq datasets from lean, obese, and T2DM patients, we reveal elevated mitochondrial reactive oxygen species (ROS) pathway and NF-κB signaling with altered fatty acid metabolism in T2DM adipocytes. Mice with adipose-specific deletion of mitochondrial redox Trx2 develop hyperglycemia, hepatic insulin resistance, and hepatic steatosis. Trx2-deficient WAT exhibited excessive mitophagy, increased inflammation, and lipolysis. Mechanistically, mitophagy was induced through increasing ROS generation and NF-κB–dependent accumulation of autophagy receptor p62/SQSTM1, which recruits damaged mitochondria with polyubiquitin chains. Importantly, administration of ROS scavenger or NF-κB inhibitor ameliorates glucose and lipid metabolic disorders and T2DM progression in mice. Taken together, this study reveals a previously unrecognized mechanism linking mitophagy-mediated adipose inflammation to T2DM with hepatic insulin resistance.
Graphical Abstract
Related collections
Most cited references69
- Record: found
- Abstract: found
- Article: not found
Mechanisms of Insulin Action and Insulin Resistance
- Record: found
- Abstract: found
- Article: not found
Crosstalk of reactive oxygen species and NF-κB signaling.
- Record: found
- Abstract: found
- Article: not found