- Record: found
- Abstract: found
- Article: found
Mutant ASXL1 cooperates with BAP1 to promote myeloid leukaemogenesis
Read this article at
Abstract
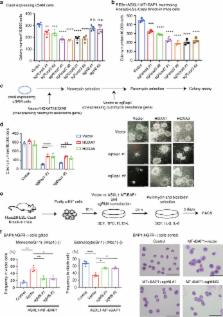
ASXL1 mutations occur frequently in myeloid neoplasms and are associated with poor prognosis. However, the mechanisms by which mutant ASXL1 induces leukaemogenesis remain unclear. In this study, we report mutually reinforcing effects between a C-terminally truncated form of mutant ASXL1 (ASXL1-MT) and BAP1 in promoting myeloid leukaemogenesis. BAP1 expression results in increased monoubiquitination of ASXL1-MT, which in turn increases the catalytic function of BAP1. This hyperactive ASXL1-MT/BAP1 complex promotes aberrant myeloid differentiation of haematopoietic progenitor cells and accelerates RUNX1-ETO-driven leukaemogenesis. Mechanistically, this complex induces upregulation of posterior HOXA genes and IRF8 through removal of H2AK119 ubiquitination. Importantly, BAP1 depletion inhibits posterior HOXA gene expression and leukaemogenicity of ASXL1-MT-expressing myeloid leukemia cells. Furthermore, BAP1 is also required for the growth of MLL-fusion leukemia cells with posterior HOXA gene dysregulation. These data indicate that BAP1, which has long been considered a tumor suppressor, in fact plays tumor-promoting roles in myeloid neoplasms.
Abstract
ASXL1 gene is often mutated in myeloid malignancies. Here, the authors show that mutant ASXL1 and BAP1 are in a positive feedback loop such that BAP1 induces monoubiquitination of mutant ASXL1, which in turn enhances BAP1 activity to potentiate myeloid transformation via HOXA clusters and IRF8.
Related collections
Most cited references43
- Record: found
- Abstract: found
- Article: not found
Clinical effect of point mutations in myelodysplastic syndromes.
- Record: found
- Abstract: found
- Article: not found
Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB.
- Record: found
- Abstract: found
- Article: not found