- Record: found
- Abstract: found
- Article: found
DICER1 mutation and pituitary prolactinoma
Summary
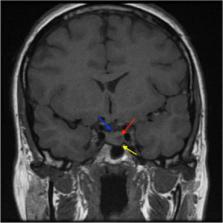
A young woman carrying germline DICER1 mutation was discovered to have a pituitary microprolactinoma when she became amenorrhoic. The mutation was identified as a result of family screening following the early death of the patient’s daughter with ovarian cancer. The patient was in follow-up screening for thyroid disease, and investigations were initiated when she became amenorrhoic. MR scan revealed a 6 mm diameter pituitary microadenoma and raised prolactin. The prolactin was efficiently suppressed with low-dose cabergoline, and her menstrual cycles resumed. Dicer is an RNase enzyme, which is essential for processing small non-coding RNAs. These molecules play pleiotropic roles in regulating gene expression, by targeting mRNA sequences for degradation. DICER1 plays different roles depending on cell context, but is thought to be a functional tumour suppressor gene. Accordingly, germline mutation in one DICER1 allele is insufficient for oncogenesis, and a second hit on the other allele is required, as a result of postnatal somatic mutation. Loss of DICER1 is linked to multiple tumours, with prominent endocrine representation. Multinodular goitre is frequent, with increased risk of differentiated thyroid cancer. Rare, developmental pituitary tumours are reported, including pituitary blastoma, but not reports of functional pituitary adenomas. As DICER1 mutations are rare, case reports are the only means to identify new manifestations and to inform appropriate screening protocols.
Learning points:
-
DICER1 mutations lead to endocrine tumours.
-
DICER1 is required for small non-coding RNA expression.
-
DICER1 carriage and microprolactinoma are both rare, but here are reported in the same individual, suggesting association.
-
Endocrine follow-up of patients carrying DICER1 mutations should consider pituitary disease.
Related collections
Most cited references10
- Record: found
- Abstract: found
- Article: not found
DICER1: mutations, microRNAs and mechanisms.
- Record: found
- Abstract: found
- Article: not found
Pituitary blastoma: a pathognomonic feature of germ-line DICER1 mutations.
- Record: found
- Abstract: found
- Article: not found
Novel Genetic Causes of Pituitary Adenomas.
Author and article information
Journal
Affiliations
Author notes
Article
EDM180087
 This work is licensed under a
Creative Commons Attribution 3.0 International License.
This work is licensed under a
Creative Commons Attribution 3.0 International License.