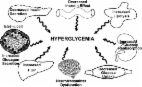
Insulin resistance in muscle and liver and β-cell failure represent the core pathophysiologic defects in type 2 diabetes. It now is recognized that the β-cell failure occurs much earlier and is more severe than previously thought. Subjects in the upper tertile of impaired glucose tolerance (IGT) are maximally/near-maximally insulin resistant and have lost over 80% of their β-cell function. In addition to the muscle, liver, and β-cell (triumvirate), the fat cell (accelerated lipolysis), gastrointestinal tract (incretin deficiency/resistance), α-cell (hyperglucagonemia), kidney (increased glucose reabsorption), and brain (insulin resistance) all play important roles in the development of glucose intolerance in type 2 diabetic individuals. Collectively, these eight players comprise the ominous octet and dictate that: 1) multiple drugs used in combination will be required to correct the multiple pathophysiological defects, 2) treatment should be based upon reversal of known pathogenic abnormalities and not simply on reducing the A1C, and 3) therapy must be started early to prevent/slow the progressive β-cell failure that already is well established in IGT subjects. A treatment paradigm shift is recommended in which combination therapy is initiated with diet/exercise, metformin (which improves insulin sensitivity and has antiatherogenic effects), a thiazolidinedione (TZD) (which improves insulin sensitivity, preserves β-cell function, and exerts antiatherogenic effects), and exenatide (which preserves β-cell function and promotes weight loss). Sulfonylureas are not recommended because, after an initial improvement in glycemic control, they are associated with a progressive rise in A1C and progressive loss of β-cell function. NATURAL HISTORY OF TYPE 2 DIABETES The natural history of type 2 diabetes has been well described in multiple populations (1 –16) (rev. in (17,18). Individuals destined to develop type 2 diabetes inherit a set of genes from their parents that make their tissues resistant to insulin (1,16,19 –24). In liver, the insulin resistance is manifested by an overproduction of glucose during the basal state despite the presence of fasting hyperinsulinemia (25) and an impaired suppression of hepatic glucose production (HGP) in response to insulin (26), as occurs following a meal (27). In muscle (19,26,28,29), the insulin resistance is manifest by impaired glucose uptake following ingestion of a carbohydrate meal and results in postprandial hyperglycemia (27). Although the origins of the insulin resistance can be traced to their genetic background (17,20), the epidemic of diabetes that has enveloped westernized countries is related to the epidemic of obesity and physical inactivity (30). Both obesity (31) and decreased physical activity (32) are insulin-resistant states and, when added to the genetic burden of the insulin resistance, place a major stress on the pancreatic β-cells to augment their secretion of insulin to offset the defect in insulin action (1,17). As long as the β-cells are able to augment their secretion of insulin sufficiently to offset the insulin resistance, glucose tolerance remains normal (33). However, with time the β-cells begin to fail and initially the postprandial plasma glucose levels and subsequently the fasting plasma glucose concentration begin to rise, leading to the onset of overt diabetes (1 –4,12,17,18,34). Collectively, the insulin resistance in muscle and liver and β-cell failure have been referred to as the triumvirate (1) (Fig. 1). The resultant hyperglycemia and poor metabolic control may cause a further decline in insulin sensitivity, but it is the progressive β-cell failure that determines the rate of disease progression. FIG. 1. Pathogenesis of type 2 diabetes: the triumvirate. Insulin resistance in muscle and liver and impaired insulin secretion represent the core defects in type 2 diabetes (1). See text for a more detailed explanation. The natural history of type 2 diabetes described above (1) is depicted by a prospective study carried out by Felber and colleagues in Lausanne, Switzerland (35) (Fig. 2). Although the study was originally cross-sectional in nature, subjects were followed up for 6 years and shown to progress from one category of glucose intolerance to the next. All subjects had a euglycemic insulin clamp to measure tissue sensitivity to insulin and an oral glucose tolerance test (OGTT) to provide an overall measure of glucose homeostasis and β-cell function. In lean subjects with normal glucose tolerance (NGT), the mean plasma glucose and insulin concentrations during the OGTT were 115 mg/dl and 62 μU/ml, while the mean rate of insulin-stimulated glucose disposal (measured with a 40 mU/m2 per min euglycemic insulin clamp) was 265 mg/m2 per min. Obesity was associated with a 29% decline in insulin sensitivity, but glucose tolerance remained perfectly normal because of the compensatory increase in insulin secretion. With time the obese NGT individuals progressed to IGT in association with a further 28% reduction in insulin sensitivity (total decrease = 57% from NGT to IGT). However, the rise in plasma glucose concentration was quite modest because of a further compensatory increase in insulin secretion. However, people with IGT are in a very precarious position. They are maximally or near-maximally insulin resistant, and their β-cells are functioning at less than maximum capacity. With time the β-cells cannot continue to produce these very large amounts of insulin and the obese IGT individual progresses to overt diabetes. The decline in glucose tolerance is associated with a marked decrease in insulin secretion without further change in insulin sensitivity (Fig. 2). This characteristic rise in insulin response to insulin resistance and hyperglycemia, followed by a subsequent decline, has been referred to as Starling's curve of the pancreas (1). This natural history of type 2 diabetes has been demonstrated in many prospective studies carried out in many diverse ethnic populations (1 –18,36,37). Although the relative contributions of insulin resistance and β-cell failure to the development of type 2 diabetes may differ in different ethnic groups (38), the onset and pace of β-cell failure determines the rate of progression of hyperglycemia. FIG. 2. Natural history of type 2 diabetes. The plasma insulin response (○) depicts the classic Starling's curve of the pancreas (1). See text for a more detailed explanation. ●, insulin-mediated glucose uptake (top panel). β-CELL FUNCTION Although the plasma insulin response to the development of insulin resistance typically is increased during the natural history of type 2 diabetes (Fig. 2), this does not mean that the β-cell is functioning normally. To the contrary, recent studies from our group have demonstrated that the onset of β-cell failure occurs much earlier and is more severe than previously appreciated. In the San Antonio Metabolism (SAM) study and the Veterans Administration Genetic Epidemiology Study (VAGES), we examined a large number of subjects with NGT (n = 318), IGT (n = 259), and type 2 diabetes (n = 201) (39 –42). All subjects had an OGTT with plasma glucose and insulin concentrations measured every 15 min to evaluate overall glucose tolerance and β-cell function and a euglycemic insulin clamp to measure insulin sensitivity. It now is recognized that simply measuring the plasma insulin response to a glucose challenge does not provide a valid index of β-cell function (43). The β-cell responds to an increment in glucose (ΔG) with an increment in insulin (ΔI) (43). Thus, a better measure of β-cell function is ΔI/ΔG. However, the β-cell also is keenly aware of the body's sensitivity to insulin and adjusts its secretion of insulin to maintain normoglycemia (33,43 –45). Thus, the gold standard for measuring β-cell function is the insulin secretion/insulin resistance (ΔI/ΔG ÷ IR), or so called disposition, index. Note that insulin resistance is the inverse of insulin sensitivity. Supplemental Fig. A1 (available in an online appendix at http://diabetes.diabetesjournals.org/cgi/content/full/db09-9028/DC1) displays the glucose area under the curve (AUC) and insulin AUC in NGT, IGT, and type 2 diabetic subjects who participated in VAGES and SAM. In the right panel, the typical inverted U-shaped or Starling's curve of the pancreas for the plasma insulin response is evident. Although subjects with IGT have an increase in the absolute plasma insulin concentration, this should not be interpreted to mean that the β-cells in these individuals are functioning normally. Figure 3 depicts the insulin secretion/insulin resistance index (ΔI/ΔG ÷ IR) in NGT, IGT, and type 2 diabetic subjects as a function of the 2-h plasma glucose concentration during the OGTT. If a 2-h plasma glucose 51%, there was a progressive decline in the log of HOMA-β. The decline in β-cell function was strongly correlated with the increase in fasting plasma glucose concentration. Studies by Butler and colleagues (93,94) have also provided additional evidence for a β-cell toxic effect for the soluble IAPP fibrils. Because amylin is secreted in a one-to-one ratio with insulin (95,96) and IAPP oligomers are toxic (89,93,94), interventions that improve insulin sensitivity, i.e., TZDs/metformin/weight loss, by leading to a reduction in insulin secretion, would be expected to preserve β-cell function on a long-term basis. Of note, rosiglitazone has been shown to protect human islets against human IAPP toxicity by a phosphatidylinositol (PI) 3-kinase–dependent pathway (97). Incretins. Abnormalities in the incretin axis have been shown to play an important role in the progressive β-cell failure of type 2 diabetes. GLP-1 and glucose-dependent insulinotrophic polypeptide (also called gastric inhibitory polypeptide [GIP]) account for ∼90% of the incretin effect (98 –100). In type 2 diabetes, there is a deficiency of GLP-1 (98 –100) and resistance to the action of GIP (102 –105). The deficiency of GLP-1 can be observed in individuals with IGT and worsens progressively with progression to type 2 diabetes (101). In addition to deficiency of GLP-1, there is resistance to the stimulatory effect of GLP-1 on insulin secretion (106,107). In contrast to GLP-1, plasma levels of GIP are elevated in type 2 diabetes, yet circulating plasma insulin levels are reduced (108). This suggests that there is β-cell resistance to the stimulatory effect of GIP on insulin secretion, and this, in fact, has been demonstrated (105). Of note, recent studies have shown that tight glycemic control can restore the β-cells' insulin secretory response to GIP (109). Thus, β-cell resistance to GIP is another manifestation of glucotoxicity. Because GLP-1 deficiency occurs early in the natural history of type 2 diabetes, it follows that GLP-1 replacement therapy is a logical choice to restore the deficient insulin response that is characteristic of the diabetic condition. Summary: β-cell dysfunction and development of type 2 diabetes. In summary, although insulin resistance in liver and muscle are well established early in the natural history of the disease, type 2 diabetes does not occur in the absence of progressive β-cell failure. INSULIN RESISTANCE Both the liver and muscle are severely resistant to insulin in individuals with type 2 diabetes (rev. in (1,17,18). However, when discussing insulin resistance, it is important to distinguish what is responsible for the insulin resistance in the basal or fasting state and what is responsible for the insulin resistance in the insulin-stimulated state. Liver. The brain has an obligate need for glucose and is responsible for ∼50% of glucose utilization under basal or fasting conditions (110). This glucose demand is met primarily by glucose production by the liver and to a smaller extent the kidneys (110). Following an overnight fast, the liver of nondiabetic individuals produces glucose at the rate of ∼2 mg/kg per min (1,25) (Fig. 7). In type 2 diabetic individuals, the rate of basal HGP is increased, averaging ∼2.5 mg/kg per min (1,25) (Fig. 7). In an average 80-kg person, this amounts to the addition of an extra 25–30 g of glucose to the systemic circulation every night. As shown in Fig. 7, control subjects cluster with a fasting plasma glucose concentration of ∼85–90 mg/dl, and their rate of HGP averages ∼2 mg/kg per min. In type 2 diabetic subjects, as the rate of basal HGP rises, so also does the fasting plasma glucose concentration, and these two variables are strongly correlated with an R value of 0.847 (P 50%. Thus, individuals with type 2 diabetes lack the gut factor that is responsible for augmenting hepatic glucose uptake following glucose ingestion. FIG. 10. Hepatic glucose uptake in nondiabetic and diabetic (DIAB) subjects as a function of plasma glucose and insulin concentrations and route of glucose administration (170 –174). Summary: pathogenesis. In summary, impaired insulin secretion, decreased muscle glucose uptake, increased HGP, and decreased hepatic glucose uptake all contribute to the glucose intolerance in type 2 diabetic individuals. DYSHARMONIOUS QUARTET (SUPPLEMENTAL FIG. A4) The last decade has taught us that the fat cell also plays a pivotal role in the pathogenesis of type 2 diabetes. Collectively, the fat cell and his three friends—the muscle, liver, and β-cell—comprise the harmonious quartet, or perhaps more appropriately, the dysharmonious quartet, since together they sing a very bad tune for the diabetic patient. Considerable evidence implicates deranged adipocyte metabolism and altered fat topography in the pathogenesis of glucose intolerance in type 2 diabetes (17,26,68,127,175 –178): 1) Fat cells are resistant to insulin's antilipolytic effect, leading to day-long elevation in the plasma FFA concentration (26,140,175 –179). 2) Chronically increased plasma FFA levels stimulate gluconeogenesis (180 –182), induce hepatic/muscle insulin resistance (142,183 –185), and impair insulin secretion (24,186). These FFA-induced disturbances are referred to as lipotoxicity. 3) Dysfunctional fat cells produce excessive amounts of insulin resistance–inducing, inflammatory, and atherosclerotic-provoking adipocytokines and fail to secrete normal amounts of insulin-sensitizing adipocytokines such as adiponectin (175,176). 4) Enlarged fat cells are insulin resistant and have diminished capacity to store fat (187,188). When adipocyte storage capacity is exceeded, lipid “overflows” into muscle, liver, and β-cells, causing muscle/hepatic insulin resistance and impaired insulin secretion (rev. in (175,176). Lipid can also overflow into arterial vascular smooth cells, leading to the acceleration of atherosclerosis. Using 14C-palmitate in combination with the insulin clamp technique, Groop et al. (26) demonstrated that the antilipolytic effect of insulin was markedly impaired in lean type 2 diabetic subjects, as well as in obese nondiabetic subjects (140). In both type 2 diabetic (supplemental Fig. A5) and obese nondiabetic subjects, the ability of insulin to suppress the plasma FFA concentration and inhibit FFA turnover is significantly impaired compared with lean normal glucose tolerant control subjects at all plasma insulin concentrations spanning the physiological and pharmacological range. Many investigators, including Boden, Shulman, and ourselves (181,182,185,189), have shown that a physiological elevation in the plasma FFA concentration stimulates HGP and impairs insulin-stimulated glucose uptake in liver (190) and muscle (151,182 –185,189 –194). As discussed earlier, we and others (24,186) have also shown that elevated plasma FFA levels inhibit insulin secretion. Many years ago, Professor Philip Randle (195) described his now famous cycle of substrate competition, whereby elevated FFA oxidation in muscle reciprocally impaired glucose oxidation. Although there clearly is substrate competition between FFA and glucose with respect to oxidative metabolism (196,197), FFAs have been shown to have independent effects to inhibit glycogen synthase (198,199) and both glucose transport and glucose phosphorylation (192,200). More recently, we have examined the effect of a 4-h lipid versus saline infusion on the insulin signal transduction system in healthy lean normal glucose tolerant subjects (201). Lipid was infused at three rates (30, 60, and 90 ml/h) to cause a physiological and pharmacological elevation in the plasma FFA concentration. During the saline control study, insulin increased whole-body glucose metabolism from 2.7 to 10.8 mg · kg−1 · min−1. Lipid infusion caused a dose-response decline in insulin-stimulated whole-body glucose disposal (by 22, 30, and 34%, respectively), which primarily reflects muscle. Compared with the saline control study, lipid infusion caused a dose-response inhibition of muscle insulin receptor tyrosine phosphorylation, IRS-1 tyrosine phosphorylation, PI 3-kinase activity, and Akt serine phosphorylation (Fig. 11). FIG. 11. Effect of lipid infusion to cause a physiological-pharmacological elevation in plasma FFA concentration on insulin signal transduction in healthy nondiabetic subjects (201). PY, phosphorylation. After fatty acids enter the cell, they can be converted to triglycerides, which are inert, or to toxic lipid metabolites such as fatty acyl CoAs, diacylglycerol, and ceramide. Using magnetic resonance spectroscopy, we quantitated intramyocellular triglyceride content in healthy normal glucose tolerant and type 2 diabetic subjects and demonstrated that muscle lipid content was significantly increased in the diabetic group (R.A.D., unpublished data). Similar results have been reported by Petersen et al. (202). Fatty acyl CoAs, which are known to inhibit insulin signaling (203,204), were also significantly increased in muscle in diabetic subjects (205,206). Diabetic subjects were treated with pioglitazone, which increases the expression of peroxisome proliferator–activated γ coactivator 1 (PGC-1) (207). PGC-1 is the master regulator of mitochondrial biogenesis and augments the expression of multiple genes involved in mitochondrial oxidative phosphorylation (208 –210). Pioglitazone reduced the intramyocellular lipid and fatty acyl CoA concentrations, and the decrement in muscle fatty acyl CoA content was closely related to the improvement in insulin-stimulated muscle glucose disposal (205). When we reduced the intramyocellular fatty acyl CoA content with acipimox, a potent inhibitor of lipolysis, a similar improvement in insulin-mediated glucose disposal was noted (206). Increased intramyocellular levels of diacylglycerol (194,211) and ceramides (212,213) have also been demonstrated in type 2 diabetic and obese nondiabetic subjects and shown to be related to the insulin resistance and impaired insulin signaling in muscle. Most recently, we demonstrated that a 48-h lipid infusion, designed to increase the plasma FFA concentration ∼1.5- to 2.0-fold, inhibited the expression of PGC1α, PGC1β, PDHA1, and multiple mitochondrial genes involved in oxidative phosphorylation in muscle (214), thus mimicking the pattern of gene expression observed in type 2 diabetic subjects and in the normal glucose tolerant, insulin-resistant offspring of two type 2 diabetic parents (215,216). Most recently, we examined the effect of palmitoyl carnitine on ATP synthesis in mitochondria isolated from muscle of normal glucose tolerant subjects (217). Low concentrations of palmitoyl carnitine (1–4 μmol/l) augmented ATP synthesis. However, palmitoyl carnitine concentrations >4 μmol/l were associated with marked inhibition of ATP synthesis and a decrease in the inner mitochondrial membrane potential, which provides the electromotive driving force for electron transport. Collectively, these findings provide strong support for lipotoxicity and adipocyte insulin resistance in the pathogenesis of type 2 diabetes. QUINTESSENTIAL QUINTET Although the fat cell is a worthy member of the dysharmonious quartet, the time has arrived to expand the playing field to include the gastrointestinal tissues as the fifth member of the quintessential quintet. Glucose ingestion elicits a much greater insulin response than an intravenous glucose infusion that mimics the plasma glucose concentration profile observed with oral glucose (98 –100). The great majority (>99%) of this incretin effect can be explained by two hormones: GLP-1 and GIP (98 –100). As discussed earlier, GLP-1 secretion by the L-cells of the distal small intestine is deficient (98 –100), while GIP secretion by the K-cells of the more proximal small intestine is increased, but there is resistance to the stimulatory effect of GIP on insulin secretion (102 –105). GLP-1 also is a potent inhibitor of glucagon secretion (98 –100), and the deficient GLP-1 response contributes to the paradoxical rise in plasma glucagon secretion and impaired suppression of HGP that occurs after ingestion of a mixed meal (218). Clearly, the gut is a major endocrine organ and contributes to the pathogenesis of type 2 diabetes. Studies from our laboratory have demonstrated that in healthy normal glucose tolerant subjects, approximately one-half of the suppression of HGP following a mixed meal is secondary to inhibition of glucagon secretion, the other one-half is secondary to the increase in insulin secretion, and the insulin-to-glucagon ratio correlated strongly with the suppression of HGP during the meal (218). These studies also demonstrated that a large amount of the ingested glucose load did not appear in the systemic circulation, consistent with previous studies from our laboratory (28,170 –172). This could have been the result of delayed gastric emptying, a known effect of exenatide, or an increase in splanchnic (primarily reflects liver) glucose uptake. To examine this question more directly, type 2 diabetic subjects received a 6-h meal tolerance test with the double tracer technique (1-14C-glucose orally and 3-3H-glucose intravenously) before and after 2 weeks of exenatide treatment (219). Exenatide was not given on the day of the study. The ingested glucose load was labeled with acetaminophen to follow gastric empting. Exenatide significantly reduced both the fasting and postprandial plasma glucose levels following ingestion of the meal compared with the baseline study performed prior to exenatide. The increment in insulin secretory rate divided by the increment in plasma glucose concentration increased more than twofold, demonstrating a potent stimulatory effect of exenatide on β-cell function. The increase in insulin secretion, in concert with a decline in glucagon release, led to a significant reduction in HGP following ingestion of the mixed meal. Gastric emptying was unaltered by exenatide, since the last dose of exenatide was administered more than ∼16 h prior to the meal. Neither splanchnic nor peripheral tissue glucose uptake was significantly altered. Thus, the primary effect of exenatide to improve glucose tolerance is related to the incretin's suppressive effect on HGP. Most recently, Cherrington (220) and Bergman (221) and colleagues have presented evidence in support of an effect of GLP-1 to enhance hepatic glucose uptake of ingested glucose in dogs. SETACEOUS SEXTET The sixth member, who establishes the setaceous sextet, is the pancreatic α-cell. Many groups, dating back to the 1970s, have demonstrated that the basal plasma glucagon concentration is elevated in type 2 diabetic individuals (119 –121,222 –224). The important contribution of elevated fasting plasma glucagon levels to the increased basal rate of HGP in type 2 diabetic individuals was provided by Baron et al. (122). Compared with control subjects, diabetic individuals had a markedly elevated rate of basal HGP, which correlated closely with the increase in fasting plasma glucagon concentration. Following somatostatin infusion, plasma glucagon levels declined by 44% in association with a 58% decrease in basal HGP. These results conclusively demonstrate the pivotal role of hyperglucagonemia in the pathogenesis of fasting hyperglycemia in type 2 diabetes. There also is evidence that the liver may be hypersensitive to the stimulatory effect of glucagon in hepatic gluconeogenesis (120). In summary, drugs that inhibit glucagon secretion or block the glucagon receptor are likely to be effective in treating patients with type 2 diabetes. One such example is exenatide (225), but glucagon receptor antagonists also have been shown to be effective (226). SEPTICIDAL SEPTET The next, and most recent member, implicated in the pathogenesis of type 2 diabetes is the kidney who along with the muscle, liver, α-cell, β-cell, adipocyte, and gut, forms the septicidal septet. The kidney filters ∼162 g ([glomerular filtration rate = 180 l/day] × [fasting plasma glucose = 900 mg/l]) of glucose every day. Ninty percent of the filtered glucose is reabsorbed by the high capacity SGLT2 transporter in the convoluted segment of the proximal tubule, and the remaining 10% of the filtered glucose is reabsorbed by the SGLT1 transporter in the straight segment of the descending proximal tubule (227). The result is that no glucose appears in the urine. In animal models of both type 1 and type 2 diabetes, the maximal renal tubular reabsorptive capacity, or Tm, for glucose is increased (228 –230). In humans with type 1 diabetes, Mogensen et al. (231) have shown that the Tm for glucose is increased. In human type 2 diabetes, the Tm for glucose has not been systematically examined. No studies in either type 1 or type 2 diabetic individuals have examined the splay in the glucose titration curve in humans. However, cultured human proximal renal tubular cells from type 2 diabetic patients demonstrate markedly increased levels of SGLT2 mRNA and protein and a fourfold increase in the uptake of α-methyl-d-glucopyranoside (AMG), a nonmetabolizeable glucose analog (232) (Fig. 12). FIG. 12. SGLT 2 transporter mRNA (left) and protein (middle) and glucose transport (α-methyl-d-glucopyranoside) (right) are increased in cultured renal proximal tubular epithelial cells of individuals with type 2 diabetes (T2DM) versus nondiabetic subjects (CON) (232). These observations have important clinical implications. Thus, an adaptive response by the kidney to conserve glucose, which is essential to meet the energy demands of the body, especially the brain and other neural tissues, which have an obligate need for glucose, becomes maladaptive in the diabetic patient. Instead of dumping glucose in the urine to correct the hyperglycemia, the kidney chooses to hold on to the glucose. Even worse, the ability of the diabetic kidney to reabsorb glucose appears to be augmented by an absolute increase in the renal reabsorptive capacity for glucose. In summary, the development of medications that inhibit renal proximal tubular glucose reabsorption provides a rational approach to the treatment of type 2 diabetes (227). OMINOUS OCTET (FIG. 13) The last, and perhaps most important, player to be implicated in the pathogenesis of type 2 diabetes is the brain, which, along with his seven companions, forms the ominous octet. It is abundantly clear that the current epidemic of diabetes is being driven by the epidemic of obesity (207,233). Porte and colleagues (234 –237) were among the first to demonstrate that, in rodents, insulin was a powerful appetite suppressant. Obese individuals, both diabetic and nondiabetic, are characterized by insulin resistance and compensatory hyperinsulinemia. Nonetheless, food intake is increased in obese subjects despite the presence of hyperinsulinemia, and one could postulate that the insulin resistance in peripheral tissues also extends to the brain. FIG. 13. The ominous octet. See text for a more detailed explanation. Our laboratory has attempted to address the issue of impaired appetite regulation by insulin in obese subjects using functional magnetic resonance imaging (MRI) to examine the cerebral response to an ingested glucose load (238). After glucose ingestion, two hypothalamic areas with consistent inhibition were noted: the lower posterior hypothalamus, which contains the ventromedial nuclei, and the upper posterior hypothalamus, which contains the paraventricular nuclei. In both of these hypothalamic areas, which are key centers for appetite regulation, the magnitude of the inhibitory response following glucose ingestion was reduced in obese, insulin-resistant, normal glucose tolerant subjects, and there was a delay in the time taken to reach the maximum inhibitory response, even though the plasma insulin response was markedly increased in the obese group. Whether the impaired functional MRI response in obese subjects contributes to or is a consequence of the insulin resistance and weight gain remains to be determined. Nonetheless, these results suggest that the brain, like other organs (liver, muscle, and fat) in the body, may be resistant to insulin. Studies by Obici et al. (239,240) in rodents have also provided evidence for cerebral insulin resistance leading to increased HGP and reduced muscle glucose uptake. IMPLICATIONS FOR THERAPY The preceding review of the pathophysiology of type 2 diabetes has important therapeutic implications (Table 1). First, effective treatment of type 2 diabetes will require multiple drugs used in combination to correct the multiple pathophysiological defects. Second, the treatment should be based upon known pathogenic abnormalities and NOT simply on the reduction in A1C. Third, therapy must be started early in the natural history of type 2 diabetes, if progressive β-cell failure is to be prevented. TABLE 1 Pathogenesis of type 2 diabetes: implications for therapy 1) Effective treatment of type 2 diabetes requires multiple drugs used in combination to correct multiple pathophysiological defects. 2) Treatment should be based on known pathogenic abnormalities and not simply on reduction of A1C. 3) Therapy must be started early in the natural history of type 2 diabetes to prevent progressive β-cell failure. Let us now examine the current therapeutic options as they relate to four of the key pathophysiological derangements present in type 2 diabetes (Fig. 14). At the level of the liver, we have shown that both metformin (241 –243) and the TZDs (175,244 –252) are potent insulin sensitizers and inhibit the increased rate of hepatic gluconeogenesis (220,221) that is characteristic of type 2 diabetic patients. In muscle, TZDs are potent insulin sensitizers (244 –252), whereas metformin is a very weak insulin sensitizer (241,243,253). Since the TZDs work through the classic insulin signaling pathway (150,254), whereas metformin works through the AMP kinase pathway (255,256), combination therapy with a TZD plus metformin gives a completely additive effect to reduce the A1C (257 –265), and hypoglycemia is not encountered because these drugs are insulin sensitizers and do not augment insulin secretion. In adipose tissue, the TZDs are also excellent insulin sensitizers and are potent inhibitors of lipolysis (263). TZDs also effectively mobilize fat out of muscle, liver, and β-cell, thereby ameliorating lipotoxicity (175,176,205,264 –267). FIG. 14. Treatment of type 2 diabetes: a therapeutic approach based upon pathophysiology. See text for a more detailed explanation. At the level of the β-cell, only the TZDs conclusively have been shown to improve and preserve β-cell function (75,268) and demonstrate durability of control (167,168,260, 268 –272). There is also evidence that the GLP-1 analogs can preserve β-cell function on a long-term basis (273 –275). Nonetheless, the two most commonly prescribed drugs in the U.S. and throughout the world are the sulfonylureas and metformin, and neither of these drugs exerts any significant protective effect on the β-cell. This is a major concern, since progressive β-cell failure is the primary pathogenic abnormality responsible for the development of overt diabetes and the progressive rise in A1C (Fig. 2 and supplemental Fig. A1). Sulfonylureas and metformin. Professor Robert Turner, in the UK Prospective Diabetes Study (UKPDS), was the first to conclusively show that sulfonylureas had no protective effect on the β-cell in newly diagnosed type 2 diabetic patients over the 15-year study duration (36). After an initial drop in the A1C, sulfonylurea-treated patients experienced a progressive deterioration in glycemic control that paralleled the rise in A1C in the conventionally treated group (Fig. 15). Moreover, in the UKPDS sulfonylureas were shown not to have a significant protective effect against atherosclerotic cardiovascular complications (34), and some studies even have suggested that sulfonylureas may accelerate the atherogenic process (276,277). Similarly, metformin-treated patients in the UKPDS, after an initial decline in A1C, secondary to the biguanide's inhibitory effect on HGP, also experienced a progressive deterioration in glycemic control (Fig. 15) (278). Using HOMA-β, Professors Holman and Turner showed that the relentless rise in A1C observed with both sulfonylureas and metformin resulted from a progressive decline in β-cell function and that by 3 years ∼50% of diabetic patients required an additional pharmacological agent to maintain the A1C 1.5 years), active-comparator, or placebo-controlled studies have examined the ability of sulfonylureas to produce a durable reduction in A1C in type 2 diabetic patients. All of these studies (36,166,167, 260,268 –272) showed that, after an initial decline in A1C, a variety of sulfonylureas, including glyburide, glimepiride, and gliclazide, were associated with a progressive decline in β-cell function with an accompanying loss of glycemic control (Fig. 16). There are no exceptions to this consistent loss of glycemic control with the sulfonylureas after the initial 18 months of therapy. Thus, evidence-based medicine conclusively demonstrates that the glucose-lowering effect of the sulfonylureas is not durable and that the loss of glycemic control is associated with progressive β-cell failure (36,37,166,167,268 –272,279 –283). TZDs. In contrast to the sulfonylureas, eight long-term (>1.5 years) active-comparator or double-blind placebo-controlled studies with the TZDs present a very different picture (Fig. 17) (167,168,268 –272). Thus, after an initial decline in A1C, durability of glycemic control is maintained because of the preservation of β-cell function in type 2 diabetic patients. In addition to these studies performed in type 2 diabetic patients, there are five studies in subjects with IGT demonstrating that TZDs prevent the progression of IGT to type 2 diabetes (286 –290). The DREAM (Diabetes Reduction Assessment with Ramipril and Rosiglitazone Medication) study showed a 62% decrease in the development of type 2 diabetes with rosiglitazone (287), while the ACT NOW (Actos Now for Prevention of Diabetes) study (290) showed a 81% reduction in the conversion of IGT to type 2 diabetes with pioglitazone. All five of these studies showed that, in addition to their insulin sensitizing effect, the TZDs had a major action to preserve β-cell function. In ACT NOW, the improvement in the insulin secretion/insulin resistance (disposition) index (measure of β-cell function) was shown both with the OGTT and the frequently sampled intravenous glucose tolerance test. Similar results have been demonstrated in the TRIPOD (Troglitazone In Prevention Of Diabetes) and PIPOD (Pioglitazone In Prevention Of Diabetes) studies (286,289) in which the development of diabetes in Hispanic women with a history of gestational diabetes was decreased by 52 and 62%, respectively. Many in vivo and in vitro studies with human and rodent islets have shown that TZDs exert a protective effect on β-cell function (291 –295). FIG. 17. Summary of studies examining the effect of TZDs versus placebo or versus active-comparator on A1C in type 2 diabetic subjects (167,168,260,268 –273). See text for a more detailed discussion. PIO, pioglitazone; ROSI, rosiglitazone. GLP-1 analogs. Incretins also have been shown to improve β-cell function and maintain durability of glycemic control. Bunck et al. (273) studied 69 metformin-treated type 2 diabetic patients with a mean age of 58 years and BMI of 30.5 kg/m2. Subjects received glargine insulin or exenatide to similarly reduce the A1C to 6.8%. Before and after 1 year, C-peptide secretion was evaluated with an 80-min hyperglycemic clamp. During the repeat hyperglycemic clamp performed after 1 year, both the first (0–10 min) and second (10–80 min) phases of insulin secretion were increased 1.5- and 2.9-fold, respectively, in the group treated with exenatide versus the group treated with glargine. Glargine increased by 31% the ratio of the C-peptide response during the hyperglycemic clamp performed after 1 year compared with the hyperglycemic clamp performed at baseline. In contrast, exenatide increased the ratio more than threefold, demonstrating a potent effect of this GLP-1 analog to augment β-cell function. In a 32-week double-blind, placebo-controlled study, exenatide (10 μg b.i.d.) reduced A1C by ∼1.0–1.2% and markedly decreased the postprandial rise in plasma glucose concentration while maintaining the plasma insulin response at pre-exenatide treatment levels (274). Consequently, the ΔI/ΔG ratio increased dramatically, indicating a robust effect on β-cell function. A subset of these subjects were followed-up for 3.5 years, and the decline in A1C was shown to persist (275). However, it is not known whether the subjects who did not continue in this long-term extension study had the same characteristics, i.e., level of glycemic control, etc., as those who continued to be followed for 3.5 years. In vivo studies in rodents (296,297) and in vitro studies with cultured human islets (298) have shown that exenatide can expand β-cell mass and prevent apoptosis of islets, respectively. Whether these effects to augment β-cell mass will be observed in diabetic humans remains to be determined. Irrespective of changes in β-cell mass, the studies of Bunck et al. (273) clearly document a major effect of exenatide to augment β-cell function. In addition to their effect on the β-cell, exenatide and other GLP-1 beneficially impact four other members of the ominous octet: liver (reduced HGP), α-cell (reduced glucagon secretion), gut (replacement of deficient GLP-1 response), and brain (reduced appetite with weight loss). Importantly, the stimulatory effect of exenatide on insulin secretion dissipates when normoglycemia is achieved, thereby minimizing the adverse effect of hypoglycemia. Dipeptidyl peptidase-IV inhibitors. There are no long-term studies examining the effect of the dipeptidyl peptidase-IV (DPP-IV) inhibitors on β-cell function. However, in short-term studies, from several months to 1 year, both sitagliptin and vildagliptin (98,99,299,300) reduce the postprandial plasma glucose concentration while maintaining the plasma insulin response, indicating a positive effect on β-cell function. Whether this enhancement in insulin secretion will be translated into preservation of β-cell function on a long-term basis remains to be determined. The DPP-IV inhibitors also decrease glucagon secretion, and in concert with the rise in plasma insulin, this leads to a reduction in basal HGP (301). Hypoglycemia does not occur with the DPP-IV inhibitors, but they do not suppress appetite or cause weight loss. Summary. The introduction of the TZDs and GLP-1 analogs into the diabetes market place and their potential to preserve β-cell function offer a new therapeutic approach to the treatment of type 2 diabetes. ADA ALGORITHM FOR TREATMENT OF TYPE 2 DIABETES The ADA algorithm for the treatment of type 2 diabetes advocates a stepwise therapeutic approach that is based upon reduction in the plasma glucose concentration and NOT upon known pathophysiological disturbances (49). It dictates the initiation of therapy with lifestyle modification plus metformin to achieve an A1C < 7.0% (Fig. 18). If the goal is not reached or if secondary failure occurs, the ADA algorithm suggests one of three options: 1) First is the addition of basal insulin, an option unlikely to be chosen by primary care physicians or most endocrinologists in the U.S. and unlikely to achieve the desired level of glycemic control based upon well-designed studies by experts in the field of insulin therapy (302 –308). Moreover, all of these insulin-based add-on studies have been associated with a high incidence of hypoglycemia and major weight gain (range 4.2–19.2 lbs, mean 8.5 lbs within 6–12 months or less) (Fig. 19). 2) Second is the addition of a TZD, but this option is unlikely to be chosen because of the concerns raised in the ADA algorithm about this class of drugs. Thus, the ADA algorithm basically guides the physician to select a sulfonylurea as the choice for a second antidiabetic agent. Moreover, third party reimbursers like this option because sulfonylureas are inexpensive. Neither the GLP-1 analogs nor the DPP-4 inhibitors are included as an option in the ADA algorithm (49). Since neither the sulfonylureas nor metformin exerts any effect to preserve β-cell function (see previous discussion and Fig. 16), the 20% of β-cell function that was present at the time of diagnosis of diabetes (40 –42) will largely have been lost by the time that combined sulfonylurea/metformin therapy has failed, and the majority of these patients will require insulin treatment. Insulin therapy is difficult for most primary care physicians, and even in the hands of experienced endocrinologists it is not easy to achieve and maintain an A1C <7%—let alone <6.5%—without significant hypoglycemia and weight gain (302 –308). Moreover, it is unclear why one would initiate insulin before exenatide, since insulin rarely decreases the A1C to <7.0% and is associated with significant weight gain and hypoglycemia (302 –308) (Fig. 19). Most recently, an ADA Consensus Statement has significantly revised the ADA therapeutic algorithm (309). A two-tier approach is advocated, and sulfonylureas have been elevated into the first tier and are to be used if diet/exercise plus metformin fail to reduce the A1C to <7.0% (Fig. 20). From the pathophysiological standpoint, this represents a major step backward, since an overwhelming body of evidence-based medicine (Fig. 16) conclusively demonstrates that sulfonylureas do not preserve β-cell function and do not achieve durability of glycemic control. Although this algorithm is not the official policy statement of ADA, it is likely to be interpreted as such by most third-party payers. FIG. 18. ADA algorithm for the treatment of type 2 diabetes (49). See text for a more detailed explanation. SU, sulfonylurea. FIG. 19. Effect of insulin (Ins) and exenatide on A1C and body weight in type 2 diabetic subjects (302 –308). FIG. 20. ADA consensus statement algorithm on the treatment of type 2 diabetes. As indicated, this does not represent the official statement of ADA (49). See text for a detailed discussion (309). Exen, exenatide; PIO, pioglitazone; SU, sulfonylurea. PATHOPHYSIOLOGICAL-BASED ALGORITHM An alternate therapeutic algorithm is based upon known pathophysiological disturbances in type 2 diabetes (Fig. 21). This algorithm provides a more rational approach and is more likely to produce a durable long-term effect. This algorithm initiates treatment with lifestyle modification plus triple combination therapy with drugs known to improve insulin sensitivity (TZDs and metformin) and, most importantly, with drugs that have been shown to preserve β-cell function (TZDs and exenatide) (Fig. 21). Further, a more rational goal of therapy should be an A1C <6.0%, since the DPP has taught us that as many as 12% of individuals with IGT and an A1C of 6.0% already have background diabetic retinopathy. FIG. 21. Pathophysiological-based algorithm: treatment of type 2 diabetes based upon pathophysiology. See text for a detailed discussion. Comparison of the stepwise ADA algorithm with the combination pathophysiological-based algorithm is shown in Fig. 22. Many studies, including the UKPDS, have shown that stepped metformin/sulfonylurea therapy does not achieve durable glycemic control. Conversely, the TZDs and the GLP-1 analogs, when used as monotherapy, each have been shown to have a more durable effect. When used in combination, if anything, one would hypothesize an even more durable effect on β-cell function and reduction in A1C, although this remains to be proven. Neither the sulfonylureas nor metformin has been shown to preserve β-cell function. In contrast, both the TZDs and exenatide have been shown to preserve β-cell function. Hypoglycemia is common with the sulfonylureas and insulin, and this prohibits the achievement of the optimal A1C goal of 6.0%, let alone an A1C <7.0% (the ADA-recommended goal). In contrast, hypoglycemia is uncommon with the insulin sensitizers and GLP-1 analogs, allowing the physician to titrate these drugs to maximum doses to reduce the A1C <6.0%. Lastly, weight gain is common with sulfonylurea and insulin therapy, whereas weight loss is the norm with exenatide, and exenatide blocks the weight gain that is associated with the TZDs. FIG. 22. Comparison of the ADA and pathophysiological-based algorithms. See text for a detailed discussion. Summary: Treatment. Although this paradigm shift, which is based upon pathophysiology, represents a novel approach to the treatment of type 2 diabetes, it is substantiated by a vast body of basic scientific and clinical investigational studies. Because this algorithm is based upon the reversal of known pathophysiological defects, it has a high probability of achieving durable glycemic control. If the plasma glucose concentration can be maintained within the normal nondiabetic range, the microvascular complications of the disease, which are costly to treat and associated with major morbidity and mortality, can be prevented. Most importantly, this will enhance the quality of life for all diabetic patients. Supplementary Material Online-Only Appendix