- Record: found
- Abstract: found
- Article: found
Antimicrobial resistance and population genomics of multidrug-resistant Escherichia coli in pig farms in mainland China
Read this article at
Abstract
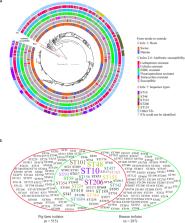
The expanding use of antimicrobials in livestock is an important contributor to the worldwide rapid increase in antimicrobial resistance (AMR). However, large-scale studies on AMR in livestock remain scarce. Here, we report findings from surveillance of E. coli AMR in pig farms in China in 2018–2019. We isolated E. coli in 1,871 samples from pigs and their breeding environments, and found AMR in E. coli in all provinces in mainland China. We detected multidrug-resistance in 91% isolates and found resistance to last-resort drugs including colistin, carbapenems and tigecycline. We also identified a heterogeneous group of O-serogroups and sequence types among the multidrug-resistant isolates. These isolates harbored multiple resistance genes, virulence factor-encoding genes, and putative plasmids. Our data will help to understand the current AMR profiles of pigs and provide a reference for AMR control policy formulation for livestock in China.
Abstract
Use of antimicrobials in livestock contributes to development of antimicrobial resistance but there are few large-scale surveillance studies. Here, the authors describe E. coli surveillance in pig farms in China, reporting high levels of multidrug-resistance across all mainland provinces.
Related collections
Most cited references63

- Record: found
- Abstract: found
- Article: found
FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments

- Record: found
- Abstract: found
- Article: found
The RAST Server: Rapid Annotations using Subsystems Technology
- Record: found
- Abstract: found
- Article: found