- Record: found
- Abstract: found
- Article: found
Manumycin A corrects aberrant splicing of Clcn1 in myotonic dystrophy type 1 (DM1) mice
Read this article at
Abstract
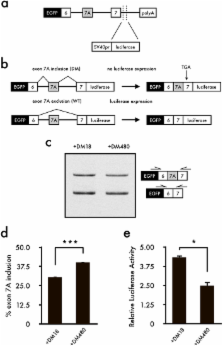
Myotonic dystrophy type 1 (DM1) is the most common muscular dystrophy in adults and as yet no cure for DM1. Here, we report the potential of manumycin A for a novel DM1 therapeutic reagent. DM1 is caused by expansion of CTG repeat. Mutant transcripts containing expanded CUG repeats lead to aberrant regulation of alternative splicing. Myotonia (delayed muscle relaxation) is the most commonly observed symptom in DM1 patients and is caused by aberrant splicing of the skeletal muscle chloride channel ( CLCN1) gene. Identification of small-molecule compounds that correct aberrant splicing in DM1 is attracting much attention as a way of improving understanding of the mechanism of DM1 pathology and improving treatment of DM1 patients. In this study, we generated a reporter screening system and searched for small-molecule compounds. We found that manumycin A corrects aberrant splicing of Clcn1 in cell and mouse models of DM1.
Related collections
Most cited references32
- Record: found
- Abstract: found
- Article: not found
The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants.
- Record: found
- Abstract: found
- Article: not found
Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9.
- Record: found
- Abstract: found
- Article: not found