- Record: found
- Abstract: found
- Article: found
A PCR protocol to establish standards for routine mycoplasma testing that by design detects over ninety percent of all known mycoplasma species
Read this article at
Summary
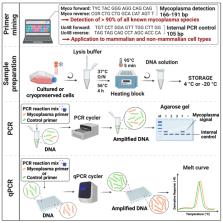
Mycoplasma infection leads to false and non-reproducible scientific data and poses a risk to human health. Despite strict guidelines calling for regular mycoplasma screening, there is no universal and widely established standard procedure. Here, we describe a reliable and cost-effective PCR method that establishes a universal protocol for mycoplasma testing. The applied strategy utilizes ultra-conserved eukaryotic and mycoplasma sequence primers covering by design 92% of all species in the six orders of the class Mollicutes within the phylum Mycoplasmatota and is applicable to mammalian and many non-mammalian cell types. This method can stratify mycoplasma screening and is suitable as a common standard for routine mycoplasma testing.
Graphical abstract

Highlights
Abstract
Microbiology; Mycology; Sequence analysis
Related collections
Most cited references57
- Record: found
- Abstract: found
- Article: not found
MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform.
- Record: found
- Abstract: found
- Article: not found
The Human Genome Browser at UCSC

- Record: found
- Abstract: found
- Article: found