- Record: found
- Abstract: found
- Article: found
An African origin for Mycobacterium bovis
Read this article at
Abstract
Background and objectives
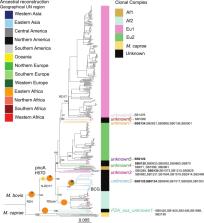
Mycobacterium bovis and Mycobacterium caprae are two of the most important agents of tuberculosis in livestock and the most important causes of zoonotic tuberculosis in humans. However, little is known about the global population structure, phylogeography and evolutionary history of these pathogens.
Methodology
We compiled a global collection of 3364 whole-genome sequences from M.bovis and M.caprae originating from 35 countries and inferred their phylogenetic relationships, geographic origins and age.
Results
Our results resolved the phylogenetic relationship among the four previously defined clonal complexes of M.bovis, and another eight newly described here. Our phylogeographic analysis showed that M.bovis likely originated in East Africa. While some groups remained restricted to East and West Africa, others have subsequently dispersed to different parts of the world.
Conclusions and implications
Our results allow a better understanding of the global population structure of M.bovis and its evolutionary history. This knowledge can be used to define better molecular markers for epidemiological investigations of M.bovis in settings where whole-genome sequencing cannot easily be implemented.
Lay summary
During the last few years, analyses of large globally representative collections of whole-genome sequences (WGS) from the human-adapted Mycobacterium tuberculosis complex (MTBC) lineages have enhanced our understanding of the global population structure, phylogeography and evolutionary history of these pathogens. In contrast, little corresponding data exists for M. bovis, the most important agent of tuberculosis in livestock. Using whole-genome sequences of globally distributed M. bovis isolates, we inferred the genetic relationships among different M. bovis genotypes distributed around the world. The most likely origin of M. bovis is East Africa according to our inferences. While some M. bovis groups remained restricted to East and West Africa, others have subsequently dispersed to different parts of the world driven by cattle movements.
Related collections
Most cited references37
- Record: found
- Abstract: found
- Article: not found
A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data.
- Record: found
- Abstract: found
- Article: not found
Time dependency of molecular rate estimates and systematic overestimation of recent divergence times.
- Record: found
- Abstract: found
- Article: found