- Record: found
- Abstract: found
- Article: found
Management of bone disease in cystinosis: Statement from an international conference
Read this article at
Abstract
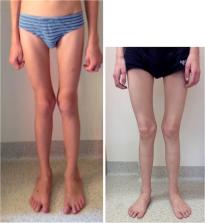
Cystinosis is an autosomal recessive storage disease due to impaired transport of cystine out of lysosomes. Since the accumulation of intracellular cystine affects all organs and tissues, the management of cystinosis requires a specialized multidisciplinary team consisting of pediatricians, nephrologists, nutritionists, ophthalmologists, endocrinologists, neurologists' geneticists, and orthopedic surgeons. Treatment with cysteamine can delay or prevent most clinical manifestations of cystinosis, except the renal Fanconi syndrome. Virtually all individuals with classical, nephropathic cystinosis suffer from cystinosis metabolic bone disease (CMBD), related to the renal Fanconi syndrome in infancy and progressive chronic kidney disease (CKD) later in life. Manifestations of CMBD include hypophosphatemic rickets in infancy, and renal osteodystrophy associated with CKD resulting in bone deformities, osteomalacia, osteoporosis, fractures, and short stature. Assessment of CMBD involves monitoring growth, leg deformities, blood levels of phosphate, electrolytes, bicarbonate, calcium, and alkaline phosphatase, periodically obtaining bone radiographs, determining levels of critical hormones and vitamins, such as thyroid hormone, parathyroid hormone, 25(OH) vitamin D, and testosterone in males, and surveillance for nonrenal complications of cystinosis such as myopathy. Treatment includes replacement of urinary losses, cystine depletion with oral cysteamine, vitamin D, hormone replacement, physical therapy, and corrective orthopedic surgery. The recommendations in this article came from an expert meeting on CMBD that took place in Salzburg, Austria, in December 2016.
Related collections
Most cited references55
- Record: found
- Abstract: found
- Article: not found
A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis.
- Record: found
- Abstract: found
- Article: not found
Vitamin D status and mortality risk in CKD: a meta-analysis of prospective studies.
- Record: found
- Abstract: found
- Article: not found