- Record: found
- Abstract: found
- Article: found
Limb girdle muscular dystrophy type 2L presenting as necrotizing myopathy
Read this article at
Abstract
Recessive mutations in the ANO5 gene, encoding anoctamin 5, cause proximal limb girdle muscular dystrophy (LGMD2L), Miyoshi-type distal myopathy (MM3) and asymptomatic hyper- CKemia.
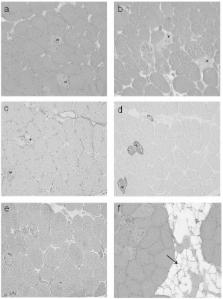
We report a woman with exertion-induced myalgia and weakness in the hip girdle manifesting at the age of 40. Creatine kinase (CK) was increased 20-fold. Histologically the dominating feature was necrotizing myopathy, but long-term immunosuppressive therapy did not change CK level or myopathic symptoms. Molecular genetic investigation led to the finding of the homozygous ANO5 c.191dupA mutation. This is a report of a muscular dystrophy due to ANO5 mutation presenting histologically as necrotizing myopathy. For this reason our finding extends the histological spectrum of myopathies due to ANO5 mutations as well as the possible differential diagnoses for necrotizing myopathy.
Related collections
Most cited references10
- Record: found
- Abstract: found
- Article: not found
Recessive mutations in the putative calcium-activated chloride channel Anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies.
- Record: found
- Abstract: found
- Article: not found
A founder mutation in Anoctamin 5 is a major cause of limb-girdle muscular dystrophy.

- Record: found
- Abstract: found
- Article: not found