- Record: found
- Abstract: found
- Article: found
Metaproteomics analysis of the functional insights into microbial communities of combined hydrogen and methane production by anaerobic fermentation from reed straw
Read this article at
Abstract
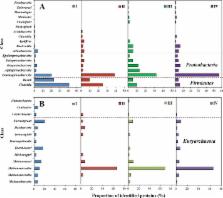
A metaproteomic approach was used to analyse the proteins expressed and provide functional evidence of key metabolic pathways in the combined production of hydrogen and methane by anaerobic fermentation (CHMP-AF) for reed straw utilisation. The functions and structures of bacteria and archaea populations show significant succession in the CHMP-AF process. There are many kinds of bacterial functional proteins, mainly belonging to phyla Firmicutes, Proteobacteria, Actinobacteria and Bacteroidetes, that are involved in carbohydrate metabolism, energy metabolism, lipid metabolism, and amino acid metabolism. Ferredoxin-NADP reductase, present in bacteria in genus Azotobacter, is an important enzyme for NADH/NAD + equilibrium regulation in hydrogen production. The archaeal functional proteins are mainly involved in methane metabolism in energy metabolism, such as acetyl-CoA decarboxylase, and methyl-coenzyme M reductase, and the acetic acid pathway exhibited the highest proportion of the total. The archaea of genus Methanosarcina in phylum Euryarchaeota can produce methane under the effect of multi-functional proteins through acetic acid, CO 2 reduction, and methyl nutrient pathways. The study demonstrates metaproteomics as a new way of uncovering community functional and metabolic activity. The combined information was used to identify the metabolic pathways and organisms crucial for lignocellulosic biomass degradation and biogas production. This also regulates the process from its protein levels and improves the efficiency of biogas production using reed straw biomass.
Related collections
Most cited references34
- Record: found
- Abstract: found
- Article: not found
Comparative analysis of bacterial community structure in the rhizosphere of maize by high-throughput pyrosequencing
- Record: found
- Abstract: found
- Article: not found
Functional metaproteome analysis of protein extracts from contaminated soil and groundwater.
- Record: found
- Abstract: found
- Article: not found