- Record: found
- Abstract: found
- Article: found
CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers
Read this article at
Abstract
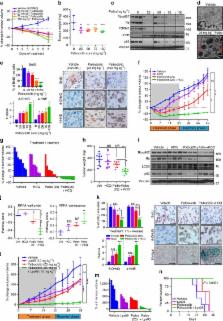
Deregulation of the cell cycle machinery is a hallmark of cancer. While CDK4/6 inhibitors are FDA approved (palbociclib) for treating advanced estrogen receptor-positive breast cancer, two major clinical challenges remain: (i) adverse events leading to therapy discontinuation and (ii) lack of reliable biomarkers. Here we report that breast cancer cells activate autophagy in response to palbociclib, and that the combination of autophagy and CDK4/6 inhibitors induces irreversible growth inhibition and senescence in vitro, and diminishes growth of cell line and patient-derived xenograft tumours in vivo. Furthermore, intact G1/S transition (Rb-positive and low-molecular-weight isoform of cyclin E (cytoplasmic)-negative) is a reliable prognostic biomarker in ER positive breast cancer patients, and predictive of preclinical sensitivity to this drug combination. Inhibition of CDK4/6 and autophagy is also synergistic in other solid cancers with an intact G1/S checkpoint, providing a novel and promising biomarker-driven combination therapeutic strategy to treat breast and other solid tumours.
Abstract
CDK4/6-Cyclin D pathway is often deregulated in cancer; therefore specific inhibitors have been developed. Here the authors show that treatment with CDK4/6 inhibitors activate autophagy in breast cancer cells; thus, combination of such inhibitors with autophagy inhibitors results in a synergistic effect on tumour growth.
Related collections
Most cited references51
- Record: found
- Abstract: found
- Article: not found
A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells.
- Record: found
- Abstract: found
- Article: not found