- Record: found
- Abstract: found
- Article: found
Comparative Analyses of the Bacterial Microbiota of the Human Nostril and Oropharynx
Read this article at
Abstract
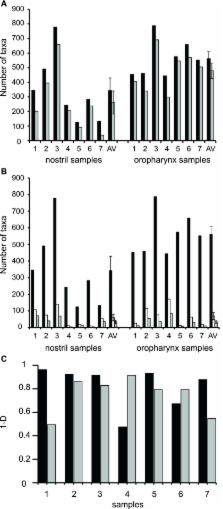
The nose and throat are important sites of pathogen colonization, yet the microbiota of both is relatively unexplored by culture-independent approaches. We examined the bacterial microbiota of the nostril and posterior wall of the oropharynx from seven healthy adults using two culture-independent methods, a 16S rRNA gene microarray (PhyloChip) and 16S rRNA gene clone libraries. While the bacterial microbiota of the oropharynx was richer than that of the nostril, the oropharyngeal microbiota varied less among participants than did nostril microbiota. A few phyla accounted for the majority of the bacteria detected at each site: Firmicutes and Actinobacteria in the nostril and Firmicutes, Proteobacteria, and Bacteroidetes in the oropharynx. Compared to culture-independent surveys of microbiota from other body sites, the microbiota of the nostril and oropharynx show distinct phylum-level distribution patterns, supporting niche-specific colonization at discrete anatomical sites. In the nostril, the distribution of Actinobacteria and Firmicutes was reminiscent of that of skin, though Proteobacteria were much less prevalent. The distribution of Firmicutes, Proteobacteria, and Bacteroidetes in the oropharynx was most similar to that in saliva, with more Proteobacteria than in the distal esophagus or mouth. While Firmicutes were prevalent at both sites, distinct families within this phylum dominated numerically in each. At both sites there was an inverse correlation between the prevalences of Firmicutes and another phylum: in the oropharynx, Firmicutes and Proteobacteria, and in the nostril, Firmicutes and Actinobacteria. In the nostril, this inverse correlation existed between the Firmicutes family Staphylococcaceae and Actinobacteria families, suggesting potential antagonism between these groups.
IMPORTANCE
The human nose and throat, though connected, contain distinct niches that are important sites of colonization by pathogenic bacteria. For many of these pathogens, colonization increases the risk of infection. Most research on the microbiota of nose and throat habitats has focused on carriage of one or a few pathogens. We hypothesized that increased knowledge of the composition of the complex bacterial communities in which these pathogens reside would provide new insights into why some individuals become colonized with pathogens, while others do not. Indeed, in the nostril microbiota of participants, there was an inverse correlation between the prevalences of the Staphylococcaceae family ( Firmicutes), whose members include important pathogens, and the Corynebacteriaceae and Propionibacteriaceae families (both Actinobacteria), whose members are more commonly benign commensals. An improved understanding of competitive bacterial colonization will increase our ability to define predispositions to pathogen carriage at these sites and the subsequent risk of infection.
Related collections
Most cited references19
- Record: found
- Abstract: found
- Article: not found
Comparative Analysis of Human Gut Microbiota by Barcoded Pyrosequencing
- Record: found
- Abstract: found
- Article: not found
Bellerophon: a program to detect chimeric sequences in multiple sequence alignments.
- Record: found
- Abstract: found
- Article: not found