- Record: found
- Abstract: found
- Article: not found
Delayed onset of the diurnal melatonin rise in patients with Huntington’s disease
Read this article at

Abstract
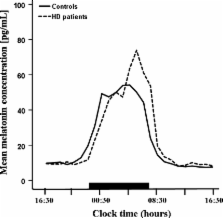
Sleep disturbances are very prevalent in Huntington’s disease (HD) patients and can substantially impair their quality of life. Accumulating evidence suggests considerable dysfunction of the hypothalamic suprachiasmatic nucleus (SCN), the biological clock, in both HD patients and transgenic mouse models of the disease. As melatonin has a major role in the regulation of sleep and other cyclical bodily activities and its synthesis is directly regulated by the SCN, we postulated that disturbed SCN function is likely to give rise to abnormal melatonin secretion in HD. Therefore, we compared 24 h melatonin secretion profiles between early stage HD patients and age-, sex- and body mass index-matched controls. Although mean diurnal melatonin levels were not different between the two groups ( p = 0.691), the timing of the evening rise in melatonin levels was significantly delayed by more than 01:30 h in HD patients ( p = 0.048). Moreover, diurnal melatonin levels strongly correlated with both motor ( r = −0.70, p = 0.036) and functional impairment ( r = +0.78, p = 0.013). These findings suggest a delayed sleep phase syndrome-like circadian rhythm disorder in early stage HD patients and suggest that melatonin levels may progressively decline with advancing disease.
Related collections
Most cited references21
- Record: found
- Abstract: found
- Article: not found
Disintegration of the sleep-wake cycle and circadian timing in Huntington's disease.
- Record: found
- Abstract: found
- Article: not found