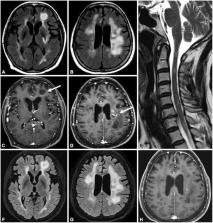
Question What is the prognostic relevance of persistent seropositivity of antibodies targeting myelin oligodendrocyte glycoprotein (MOG-IgG1) in adults after acute disseminated encephalomyelitis (ADEM)? Findings In this cohort study of 51 patients with an initial diagnosis of ADEM, relapse occurred in 8 of 9 children and 7 of 8 adults with persistent MOG-IgG1 seropositivity. By contrast, relapse occurred in 6 of 17 children and 2 of 7 adults with MOG-IgG1 seronegativity as well as 0 of 4 children and 1 of 4 adults with transient MOG-IgG1 seropositivity. Meaning Persistent MOG-IgG1 positivity after recovery from ADEM in both pediatric and adult patients is useful as a longitudinal serological biomarker predicting risk for relapsing disease. This cohort study investigates whether serostatus of autoantibodies targeting myelin oligodendrocyte glycoprotein (transient vs persistent) and titer change over time provide clinical utility in predicting the likelihood of relapse in patients after acute disseminated encephalomyelitis. Importance Recent studies have reported a higher relapse rate following an initial inflammatory demyelinating disorder in pediatric patients with persistent seropositivity of antibodies targeting myelin oligodendrocyte glycoprotein (MOG-IgG1). To date, the clinical implications of longitudinal MOG-IgG1 seropositivity using live cell assays with IgG1 secondary antibodies in adults after acute disseminated encephalomyelitis (ADEM) are unknown. Objective To determine whether MOG-IgG1 serostatus (transient vs persistent) and titer change over time provide clinical utility in predicting the likelihood of relapse after ADEM. Design, Setting, and Participants This cohort study identified patients with an initial diagnosis of ADEM evaluated at a single referral center between January 1, 1990, and October 1, 2017. Fifty-one patients were included, including 31 children and 20 adults. Longitudinal serologic testing was performed detecting autoantibodies targeting aquaporin 4 (AQP4-IgG) and MOG-IgG1 with clinically validated fluorescence-activated cell sorting assays. Patients were divided into 3 cohorts: persistent seropositivity, transient seropositivity, and seronegativity. Main Outcomes and Measures Clinical demographic characteristics, longitudinal AQP4-IgG and MOG-IgG1 serostatus, titers, relapses, use of immunotherapy, and Expanded Disability Status Scale score at follow-up. Results Of 51 patients presenting with an initial diagnosis of ADEM, 20 (39%) were adult, 24 (47%) were female, and ages ranged from 12 months to 57 years. Seventeen patients fulfilled criteria for persistent seropositivity; of those, 8 of 9 children (89%) and 7 of 8 adults (88%) had at least 1 relapse after median (range) follow-up periods of 75 (15-236) months and 39 (9-161) months, respectively. Eight patients (16%), including 4 adults, fulfilled criteria for transient seropositivity; of those, no children and 1 of 4 adults (25%) relapsed after median (range) follow-up periods of 32 (24-114) months and 16 (13-27) months, respectively. Of 24 patients with AQP4-IgG and MOG-IgG seronegativity, 6 of 17 children (35%) and 2 of 7 adults (29%) had at least 1 relapse after median (range) follow-up periods of 36 (3-203) months and 34 (15-217) months, respectively. There were only 2 patients, including 1 adult, with AQP4-IgG seropositivity, and both relapsed. The hazard ratio for relapses in those with persistent MOG-IgG1 positivity compared with AQP4-IgG and MOG-IgG1 seronegativity was 3.1 (95% CI, 1.1-8.9; P = .04) in children and 5.5 (95% CI, 1.4-22.5; P = .02) in adults. Immunotherapy was used in 5 of 9 children (56%) and 6 of 8 adults (75%) with persistent seropositivity and in 3 of 17 children (18%) and 1 of 7 adults (14%) with AQP4-IgG and MOG-IgG seronegativity. Conclusions and Relevance Relapse occurred in 15 of 17 patients (88%) with persistent MOG-IgG1 seropositivity after ADEM; only 1 patient with transient seropositivity experienced relapse. Our data extend the clinical utility of MOG-IgG1 serological testing to adult patients and highlights that longitudinal serologic evaluation of MOG-IgG1 could help predict disease course and consideration of immunotherapy.