- Record: found
- Abstract: found
- Article: found
Non-Stimulated, Agonist-Stimulated and Store-Operated Ca 2+ Influx in MDA-MB-468 Breast Cancer Cells and the Effect of EGF-Induced EMT on Calcium Entry
Read this article at
Abstract
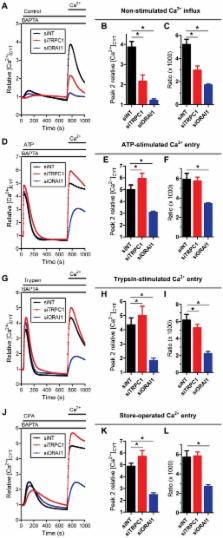
In addition to their well-defined roles in replenishing depleted endoplasmic reticulum (ER) Ca 2+ reserves, molecular components of the store-operated Ca 2+ entry pathway regulate breast cancer metastasis. A process implicated in cancer metastasis that describes the conversion to a more invasive phenotype is epithelial-mesenchymal transition (EMT). In this study we show that EGF-induced EMT in MDA-MB-468 breast cancer cells is associated with a reduction in agonist-stimulated and store-operated Ca 2+ influx, and that MDA-MB-468 cells prior to EMT induction have a high level of non-stimulated Ca 2+ influx. The potential roles for specific Ca 2+ channels in these pathways were assessed by siRNA-mediated silencing of ORAI1 and transient receptor potential canonical type 1 (TRPC1) channels in MDA-MB-468 breast cancer cells. Non-stimulated, agonist-stimulated and store-operated Ca 2+ influx were significantly inhibited with ORAI1 silencing. TRPC1 knockdown attenuated non-stimulated Ca 2+ influx in a manner dependent on Ca 2+ influx via ORAI1. TRPC1 silencing was also associated with reduced ERK1/2 phosphorylation and changes in the rate of Ca 2+ release from the ER associated with the inhibition of the sarco/endoplasmic reticulum Ca 2+-ATPase (time to peak [Ca 2+] CYT = 188.7±34.6 s (TRPC1 siRNA) versus 124.0±9.5 s (non-targeting siRNA); P<0.05). These studies indicate that EMT in MDA-MB-468 breast cancer cells is associated with a pronounced remodeling of Ca 2+ influx, which may be due to altered ORAI1 and/or TRPC1 channel function. Our findings also suggest that TRPC1 channels in MDA-MB-468 cells contribute to ORAI1-mediated Ca 2+ influx in non-stimulated cells.
Related collections
Most cited references48
- Record: found
- Abstract: found
- Article: not found
A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function.
- Record: found
- Abstract: found
- Article: not found
STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx.
- Record: found
- Abstract: found
- Article: not found