- Record: found
- Abstract: found
- Article: found
Successful treatment of myelodysplastic syndrome and Behcet colitis after allogeneic hematopoietic stem cell transplantation
letter
Min-Ha Kook
1 ,
Ho-Young Yhim
1 ,
Na-Ri Lee
1
,
2 ,
Eun-Kee Song
1
,
2 ,
Hee Sun Kim
3 ,
Chang-Yeol Yim
1
,
2 ,
Jae-Yong Kwak
1
,
2
,
02 January 2014
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
To the Editor,
Myelodysplastic syndrome (MDS) is a heterogeneous group of stem cell disorders of
unknown etiology. Behcet disease (BD) is a systemic autoimmune vasculitis also of
unknown cause. Several recent reports have suggested an association between MDS and
BD [1], possibly related to a cytogenetic abnormality such as trisomy 8 [2]. In addition,
some distinctive clinical characteristics have been identif ied that include a high
frequency of intestinal BD, generally high disease activity, and a trend toward treatment
with more potent immunosuppressive therapy [2,3]. The treatment of MDS-related Behcet
colitis is usually diff icult to manage, and many patients eventually die of infection
or hemorrhage. We herein report a case of intractable MDS-related Behcet colitis that
was successfully treated by allogeneic hematopoietic stem cell transplantation (HSCT).
A 38-year-old female was admitted to our hospital with complaints of recurrent abdominal
pain and fever. In June 2001, she was diagnosed with BD according to the diagnostic
criteria of the international BD study group [4]. She had bouts of several symptoms
compatible with BD, such as recurrent nasal ulcers, oral ulcers, genital ulcers, migrating
arthralgia, and uveitis. Total colonoscopy performed to evaluate the recurrent abdominal
pain revealed a shallow ulceration of 2 × 2 cm with irregular margins in the ileocecal
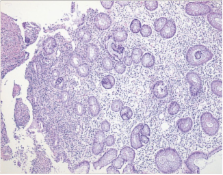
area (Fig. 1A). Pathologic examination showed chronic ulcerative inflammation with
lymphocyte infiltration compatible with Behcet colitis (Fig. 2). She was followed
closely at the rheumatology clinic and initially treated with corticosteroids alone;
however, this treatment produced no relief of symptoms. Azathioprine was added, but
she continued to experience recurrent abdominal pain. Sulfasalazine was administered
without improvement. In spite of active management of Behcet colitis, the abdominal
pain persisted in a waxing and waning manner. In April 2005, her complete blood count
(CBC) values gradually decreased, particularly the leukocyte count (2.86 × 103/µL)
and hemoglobin level (8.3 g/dL). In May 2008, the CBC showed further decreases in
the leukocyte count (1.70 × 103/µL) and hemoglobin level (5.1 g/dL), and thrombocytopenia
(90.0 × 103/µL) was detected. Bone marrow examination revealed trilineage dysplasia
with less than 5% blasts. Chromosomal analysis of bone marrow aspirates showed trisomy
8. She was diagnosed with MDS with refractory cytopenia with multilineage dysplasia.
According to the International Prognostic Scoring System, the disease was classified
as intermediate-1. For further evaluation and therapeutic options, she was referred
to our hematology clinic. She started treatment with decitabine because she did not
have a human leukocyte antigen (HLA)-matched sibling donor. However, recurrent infections
developed with complications such as neutropenic fever, pneumonia, and hepatosplenic
candidiasis. Decitabine was stopped and hematologic supportive care begun with intermittent
red cell transfusion at intervals of ~2 weeks for 15 months. We had intermittently
been searching for unrelated donors from the public blood and marrow donation program
since the diagnosis of MDS. Finally, a fully matched HLA-compatible unrelated donor
was found in January 2010. Until that time, recurrent oral ulcers, migrating arthralgia,
and abdominal pain had persisted, and she often required several days of hospitalization
to modulate the pain and associated symptoms such as diarrhea. The patient underwent
allogeneic peripheral blood stem cell transplantation with a myeloablative regimen
in April 2010. The pretransplantation conditioning regimen comprised busulfan and
cyclophosphamide. An infusion of 4.6 × 106/kg CD34+ cells was introduced. Methotrexate
and cyclosporine were used routinely for graft-versus-host disease (GVHD) prophylaxis.
On day 12, leukocyte engraftment was observed. Cyclosporine was discontinued on day
189, and abdominal pain was markedly diminished. On day 229 after transplantation,
total colonoscopy was repeated. The previous lesion was greatly improved, and only
a small shallow ulceration was observed (Fig. 1B). There was no need to treat for
Behcet colitis. At the time of this report, the patient was doing well in complete
remission of MDS without evidence of chronic GVHD and no signs or symptoms of BD.
MDS is an acquired clonal disorder of hematopoietic progenitor cells. Several recent
studies showed that an autoimmune mechanism plays an important role in the development
of MDS, and another study suggested that transformation of normal stem cells induces
an autoimmune T cell response. Although MDS and BD have been regarded as different
manifestations, the same pathophysiological processes, such as abnormal neutrophil
function, overproduction of inf lammatory cytokines, and immunological abnormalities,
are thought to be related to MDS and BD [1]. MDS-related BD shows distinctive characteristics
[2,3], notably cytogenetic abnormalities such as trisomy 8 in bone marrow cells. Especially
in Korea and Japan, bone marrow failure is frequently reported in patients with BD,
and the proportion with trisomy 8 is strikingly high (63.6% to 86.0%) [2,3]. Clinically,
BD patients with MDS have lower leukocyte counts, hemoglobin levels, and platelet
counts and significantly higher levels of serum C-reactive protein and frequencies
of intestinal BD than do BD patients without MDS. That is, patients with both BD and
MDS have higher levels of disease activity than do BD patients without MDS. As a result,
patients with MDS-related BD may be treated more frequently with corticosteroids and
other immunosuppressive agents in various combinations. However, the disease is often
refractory to these treatments. Furthermore, the control of intestinal BD in association
with MDS using immunosuppressive agents, such as prednisolone alone, is difficult,
and many patients die due to infection or hemorrhage. More encouraging are recent
reports of successful treatment of MDS-related BD using HSCT. The positive response
of the disorder to HSCT may plausibly occur through immunological reconstruction.
A very grave risk to consider in treating MDS-related BD with HSCT is the induction
of gastrointestinal GVHD. However, no reports to date have indicated that life-threatening
gastrointestinal effects develop in patients treated with HSCT, including in our patient.
In addition, patients with gastrointestinal disease before HSCT do not show an increased
risk of gastrointestinal GVHD after treatment. Autologous HSCT even improves intestinal
BD that was refractory to medical therapy [5].
In conclusion, BD may accompany MDS, possibly as an expression of a cytogenetic abnormality
such as trisomy 8. A high frequency of intestinal BD with high disease activity is
observed in MDS-related BD, and the intestinal disease may be refractory to therapy.
HSTC presents a potentially effective treatment for MDS-related BD intractable to
medical therapy without increasing the risk of gut GVHD. Further study of this treatment
approach is justified.
Related collections
Most cited references4
- Record: found
- Abstract: found
- Article: not found
Behcet's disease associated with bone marrow failure in Korean patients: clinical characteristics and the association of intestinal ulceration and trisomy 8.
Paul Yoo, Cherry Koh, D. Cha … (2008)
- Record: found
- Abstract: found
- Article: not found
Myelodysplastic syndrome complicated with inflammatory intestinal ulcers: significance of trisomy 8.
Takafumi Kawanami, Y. Hirose, Hisanori Umehara … (2005)
- Record: found
- Abstract: found
- Article: not found
Myelodysplasia-associated autoimmunity: clinical and pathophysiologic concepts.
Athanasios Tzioufas, M Voulgarelis, K Ritis … (2004)