- Record: found
- Abstract: found
- Article: found
Potential treatment of COVID-19 by inhibitors of human dihydroorotate dehydrogenase
research-article
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
The ongoing pandemic of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2),
referred to as coronavirus disease 2019 (COVID-19), has caused over 13 million infections
and over 560,000 deaths worldwide (https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports),
posing a significant threat to globally public health and economics. At present, no
efficacious antiviral drugs and vaccines have been approved for the prophylaxis or
treatment of COVID-19. Tremendous efforts have been made to develop drug and vaccine
against SARS-CoV-2. The main protease (Mpro, also called 3CLpro) is an attractive
drug target among coronaviruses, and several potent inhibitors of the SARS-CoV-2 3CLpro
together with their crystal structures in complex with the protease have been reported
(Dai et al., 2020; Jin et al., 2020; Zhang et al., 2020). While the viral RNA-dependent
RNA polymerases (RdRp) are well-known broad-spectrum antiviral drug targets, the cryo-electron
microscopy structures of the SARS-CoV-2 RdRp and its complex with remdesivir, a promising
antiviral candidate developed by Gilead Sciences, validated the efficient inhibition
of the viral RNA replication by remdesivir and provided a rational template for drug
design to combat SARS-CoV-2 infections (Gao et al., 2020; Wang et al., 2020; Yin et
al., 2020). In addition, the trimeric spike protein on the surface of SARS-CoV-2 plays
a pivotal role during the viral entry by binding to the peptidase domain of angiotensin-converting
enzyme 2 (ACE2), a host cell receptor (Yan et al., 2020). It has been revealed that
not only the receptor binding domain which is recognized by ACE2 but also the N-terminal
domain of the SARS-CoV-2 spike protein is targeting sites for therapeutic monoclonal
antibodies (Chi et al., 2020). Accordingly, both the inhibitors of 3CLpro or RdRp
and the antibodies targeting the spike protein provide potential candidates for development
of the direct-acting antiviral (DAA) drugs for the treatment of COVID-19.
In addition to DAA drugs, host-targeting antiviral (HTA) agents, targeting host proteins
required for the viral infection and replication, have advantages in overcoming drug
resistance and combating a broad spectrum of viruses including the newly emerging
virus (Ji and Li, 2020). Maraviroc, an antagonist of chemokine receptor type 5 for
HIV treatment, presents a typical HTA drug. In a remarkable study published in this
journal, Xiong et al. reported novel and potent inhibitors of human dihydroorotate dehydrogenase (DHODH)
as broad-spectrum antiviral agents against RNA viruses including SARS-CoV-2 (Xiong
et al., 2020).
Pyrimidines serve as crucial building blocks for the biosynthesis of DNA, RNA, phospholipids,
and glycoproteins, which is essential for the cell survival as well as proliferation
(Loffler et al., 2005). Human DHODH belongs to the class 2 DHODH family and is a flavin-dependent
mitochondrial enzyme catalyzing the oxidation of dihydroorotate to orotate, the fourth
step also a rate limiting step in the de novo biosynthesis of pyrimidine-based nucleotides
(Reis et al., 2017) (Fig. 1A). By consequence, DHODH is an attractive therapeutic
target for multiple diseases including cancer and autoimmune diseases (Lolli et al.,
2018; Boschi et al., 2019; Madak et al., 2019). Leflunomide and its metabolite teriflunomide,
and brequinar are well-known DHODH inhibitors and were evaluated in clinical trials
(Lolli et al., 2018). Leflunomide was approved for the therapy of rheumatoid arthritis
many years ago (Herrmann et al., 2000).
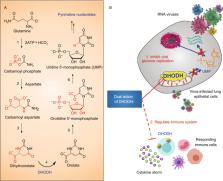
Figure 1
DHODH in the
de novo
pyrimidine biosynthesis pathway and dual action of DHODHi. (A) DHODH catalyzes the
fourth step in the de novo pyrimidine biosynthesis pathway. (B) DHODH inhibitors (DHODHi)
are broad-spectrum antivirals against RNA viruses with the dual action of inhibiting
viral genome replication and regulating the immune system
With a computer-aided hit discovery and optimization strategy, Xiong et al. identified
two novel and potent inhibitors of DHODH with a thiazole scaffold, S312 and S416 (Diao
et al., 2012; Li et al., 2015; Zhu et al., 2015). The IC50s of these two compounds
against human DHODH were 29.2 and 7.5 nmol/L, respectively, a >10-fold increase in
activity relative to the FDA-approved teriflunomide (IC50 = 307.1 nmol/L). The X-ray
crystal structure of DHODH in complex with S416 also revealed the binding mode of
two inhibitors at the ubiquinone-binding site of the enzyme. Moreover, two inhibitors
exhibited significant antiviral activities against influenza A (H1N1, H3N2 and H9N2),
Zika, Ebola, and SARS-CoV-2 in cells infected with various tested viruses, demonstrating
that DHODH inhibitors possess broad-spectrum antiviral activity by interfering the
de novo pyrimidine synthesis pathway. Low toxicities of the inhibitors suggest that
the reduced production of pyrimidine restricts virus replication but not cell growth.
Most notably, the EC50 of S416 against the viral replication in the cells infected
with SARS-CoV-2 at MOI of 0.05 is 17 nmol/L, and the resulting selectivity index (SI
= CC50/EC50) reaches 10 505.88. It is much more potent than that of teriflunomide
or brequinar and is also by far the most effective inhibitor against SARS-CoV-2 in
cells.
Another striking feature of this work is that S312 exhibited in vivo anti-influenza
efficacy equivalent to that of oseltamivir, a marketed drug for the treatment of influenza.
S312 at a dose of 5 mg/kg was also able to rescue all the influenza-infected mice
from body weight loss and death. By contrast, previous studies often showed that inhibitors
of either DHODH or the de novo pyrimidine biosynthesis pathway were ineffective against infection
in animal models. In addition, the combination administration of S312 and oseltamivir
resulted in 100% protection of the infected mice, superior to the single use of S312
or oseltamivir. S312 was also effective in the mice infected with an oseltamivir-resistant
virus and had a remarkable advantage over oseltamivir to treat the late phase of the
infectious disease. These results together demonstrated the feasibility of DHODH inhibitors
used as efficacious antivirals as well as the combination of the DHODH inhibitor with
DAA to overcome drug resistance.
As leflunomide and teriflunomide are used to treat autoimmune diseases such as rheumatoid
arthritis and multiple sclerosis by regulating lymphocytes and the release of cytokines
and chemokines, it is reasonable to conjecture that S312 and S416 would have the similar
efficacy too. As anticipated, the combination use of S312 and oseltamivir significantly
reduced the levels of IL-6, MCP-1, IL-5, KC/GRO (CXCL1), IL-2, IFN-γ, IP-10, IL-9,
TNF-α, GM-CSF, EPO, IL-12p70, MIP-3α, and IL-17A/F in the animal model. Therefore,
the DHODH inhibitors not only inhibit the viral replication but also have regulatory
roles in cytokine/chemokine production. Cytokine storm frequently occurred with patients
suffered from virus infections such as SARS-CoV and SARS-CoV-2, antiviral treatment
alone is thereby not enough and should be combined with appropriate anti-inflammatory
treatment. The DHODH inhibitors provide the ideal candidate to take both into consideration.
Taken together, this elegant work uncovers that DHODH is an attractive host target
for developing broad-spectrum antivirals which achieve the efficacy through dual mechanism
of action of antiviral and immuno-regulation (Fig. 1B), providing more therapeutic
options in response to COVID-19 as well as other emergent RNA virus infections. In
the present situation, S312 and S416, two potent inhibitors of DHODH with favorable
drug-likeness and pharmacokinetic profiles, serve as right HTAs for further evaluation
of therapeutic potential in COVID-19 treatment. Meanwhile, as a new concept for the
treatment of COVID-19, the clinical trial of leflunomide has been initiated in England
and founded by LifeArc (DEFEAT-COVID study) (https://www.lifearc.org/funding/covid-19-funding/).
Related collections
Most cited references11

- Record: found
- Abstract: found
- Article: found
Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro
Manli Wang, Ruiyuan Cao, Leike Zhang … (2020)

- Record: found
- Abstract: found
- Article: found
Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2
Renhong Yan, Yuanyuan Zhang, Yaning Li … (2020)
- Record: found
- Abstract: found
- Article: not found
Structure of Mpro from COVID-19 virus and discovery of its inhibitors
Zhenming Jin, Xiaoyu Du, Yechun Xu … (2020)