- Record: found
- Abstract: found
- Article: found
Neutrophil–lymphocyte ratio as an early new marker in AIV-H7N9-infected patients: a retrospective study
Abstract
Background: Avian AIV-H7N9 influenza progresses rapidly and has a high fatality rate. However, it lacks an early effective biomarker to predict disease severity and fatal outcomes successfully. Our study aimed to explore whether the neutrophil-to-lymphocyte ratio (NLR) taken within 24 h after admission can predict disease severity and fatality in AIV-H7N9-infected patients.
Methods: We retrospectively studied 237 AIV-H7N9-infected patients from multiple centers from 2013 to 2015. We used univariate analysis and multivariate analysis to compare clinical variables between the survival and fatal groups to evaluate the prognostic value.
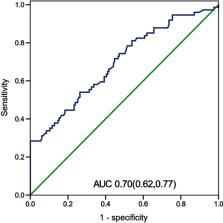
Results: The NLR taken within 24 h after admission in the fatal group was significantly higher than that in the survival group ( P<0.01). Our study found that NLR was independently associated with fatality. The area under the curve (AUC) of the NLR was 0.70, and moreover, when the NLR =19.94, the specificity was 100%, and the sensitivity was 28.4%. The fatality in the NLR ≥19.94 group was significantly increased relative to the patients with an NLR <19.94 ( P<0.05).
Conclusion: The NLR is potentially a predictive prognostic biomarker in patients infected with the AIV-H7N9 influenza virus.
Most cited references44
- Record: found
- Abstract: found
- Article: not found
Epidemiology of Human Infections with Avian Influenza A(H7N9) Virus in China


- Record: found
- Abstract: found
- Article: found