- Record: found
- Abstract: found
- Article: found
Intestinal Microbiota in Healthy Adults: Temporal Analysis Reveals Individual and Common Core and Relation to Intestinal Symptoms
Read this article at
Abstract
Background
While our knowledge of the intestinal microbiota during disease is accumulating, basic information of the microbiota in healthy subjects is still scarce. The aim of this study was to characterize the intestinal microbiota of healthy adults and specifically address its temporal stability, core microbiota and relation with intestinal symptoms. We carried out a longitudinal study by following a set of 15 healthy Finnish subjects for seven weeks and regularly assessed their intestinal bacteria and archaea with the Human Intestinal Tract (HIT)Chip, a phylogenetic microarray, in conjunction with qPCR analyses. The health perception and occurrence of intestinal symptoms was recorded by questionnaire at each sampling point.
Principal Findings
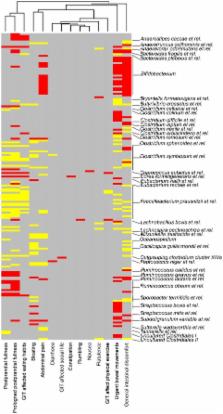
A high overall temporal stability of the microbiota was observed. Five subjects showed transient microbiota destabilization, which correlated not only with the intake of antibiotics but also with overseas travelling and temporary illness, expanding the hitherto known factors affecting the intestinal microbiota. We identified significant correlations between the microbiota and common intestinal symptoms, including abdominal pain and bloating. The most striking finding was the inverse correlation between Bifidobacteria and abdominal pain: subjects who experienced pain had over five-fold less Bifidobacteria compared to those without pain. Finally, a novel computational approach was used to define the common core microbiota, highlighting the role of the analysis depth in finding the phylogenetic core and estimating its size. The in-depth analysis suggested that we share a substantial number of our intestinal phylotypes but as they represent highly variable proportions of the total community, many of them often remain undetected.
Conclusions/Significance
A global and high-resolution microbiota analysis was carried out to determine the temporal stability, the associations with intestinal symptoms, and the individual and common core microbiota in healthy adults. The findings provide new approaches to define intestinal health and to further characterize the microbial communities inhabiting the human gut.
Related collections
Most cited references46
- Record: found
- Abstract: found
- Article: not found
Towards the human intestinal microbiota phylogenetic core.

- Record: found
- Abstract: found
- Article: found