- Record: found
- Abstract: found
- Article: found
A Biophysical Model for Analysis of Transcription Factor Interaction and Binding Site Arrangement from Genome-Wide Binding Data
Read this article at
Abstract
Background
How transcription factors (TFs) interact with cis-regulatory sequences and interact with each other is a fundamental, but not well understood, aspect of gene regulation.
Methodology/Principal Findings
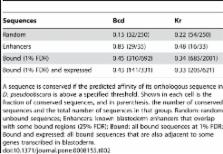
We present a computational method to address this question, relying on the established biophysical principles. This method, STAP (sequence to affinity prediction), takes into account all combinations and configurations of strong and weak binding sites to analyze large scale transcription factor (TF)-DNA binding data to discover cooperative interactions among TFs, infer sequence rules of interaction and predict TF target genes in new conditions with no TF-DNA binding data. The distinctions between STAP and other statistical approaches for analyzing cis-regulatory sequences include the utility of physical principles and the treatment of the DNA binding data as quantitative representation of binding strengths. Applying this method to the ChIP-seq data of 12 TFs in mouse embryonic stem (ES) cells, we found that the strength of TF-DNA binding could be significantly modulated by cooperative interactions among TFs with adjacent binding sites. However, further analysis on five putatively interacting TF pairs suggests that such interactions may be relatively insensitive to the distance and orientation of binding sites. Testing a set of putative Nanog motifs, STAP showed that a novel Nanog motif could better explain the ChIP-seq data than previously published ones. We then experimentally tested and verified the new Nanog motif. A series of comparisons showed that STAP has more predictive power than several state-of-the-art methods for cis-regulatory sequence analysis. We took advantage of this power to study the evolution of TF-target relationship in Drosophila. By learning the TF-DNA interaction models from the ChIP-chip data of D. melanogaster (Mel) and applying them to the genome of D. pseudoobscura (Pse), we found that only about half of the sequences strongly bound by TFs in Mel have high binding affinities in Pse. We show that prediction of functional TF targets from ChIP-chip data can be improved by using the conservation of STAP predicted affinities as an additional filter.
Related collections
Most cited references54
- Record: found
- Abstract: found
- Article: not found
DNA binding sites: representation and discovery.
- Record: found
- Abstract: found
- Article: not found
A protein interaction network for pluripotency of embryonic stem cells.

- Record: found
- Abstract: found
- Article: found