- Record: found
- Abstract: found
- Article: found
Isolation and Characterization of Numerous Novel Phages Targeting Diverse Strains of the Ubiquitous and Opportunistic Pathogen Achromobacter xylosoxidans
Read this article at
Abstract
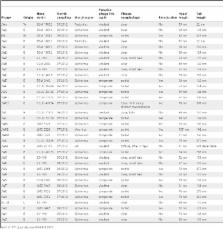
The clinical relevance of nosocomially acquired infections caused by multi-resistant Achromobacter strains is rapidly increasing. Here, a diverse set of 61 Achromobacter xylosoxidans strains was characterized by MultiLocus Sequence Typing and Phenotype MicroArray technology. The strains were further analyzed in regard to their susceptibility to 35 antibiotics and to 34 different and newly isolated bacteriophages from the environment. A large proportion of strains were resistant against numerous antibiotics such as cephalosporines, aminoglycosides and quinolones, whereas piperacillin-tazobactam, ticarcillin, mezlocillin and imipenem were still inhibitory. We also present the first expanded study on bacteriophages of the genus Achromobacter that has been so far a blank slate with respect to phage research. The phages were isolated mainly from several waste water treatment plants in Germany. Morphological analysis of all of these phages by electron microscopy revealed a broad diversity with different members of the order Caudovirales, including the families Siphoviridae, Myoviridae, and Podoviridae. A broad spectrum of different host ranges could be determined for several phages that lysed up to 24 different and in part highly antibiotic resistant strains. Molecular characterisation by DNA restriction analysis revealed that all phages contain linear double-stranded DNA. Their restriction patterns display distinct differences underlining their broad diversity.
Related collections
Most cited references63
- Record: found
- Abstract: found
- Article: not found
eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data.
- Record: found
- Abstract: found
- Article: not found
LIAN 3.0: detecting linkage disequilibrium in multilocus data. Linkage Analysis.
- Record: found
- Abstract: not found
- Article: not found