- Record: found
- Abstract: found
- Article: found
An elderly patient with 17α-hydroxylase deficiency misdiagnosed as primary aldosteronism: a case report
Read this article at
Abstract
Background
17α-hydroxylase deficiency (17OHD) is a rare autosomal recessive disorder. Aldosterone levels are usually low in patients with 17OHD. However, among the approximately 150 cases of 17OHD reported to date, aldosterone levels were not low in all cases. Therefore, some 17OHD cases may have been misdiagnosed as primary aldosteronism (PA) cases. Often before puberty, 17OHD is diagnosed because of abnormal genital morphology and menstrual irregularities. However, we report a very rare case of 17OHD in an elderly patient with a high aldosterone/renin ratio (ARR) similar to that in PA.
Case presentation
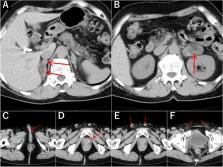
A 63-year-old Japanese woman was transferred to our medical facility for the evaluation of bilateral adrenal hypertrophy, which was incidentally discovered during an abdominal examination after cholecystectomy. The patient had hypokalemia and a high aldosterone/renin ratio. Her medical history included hypertension and right intracerebral capsular hemorrhage at the age of 30 years. Additional testing revealed low cortisol, high adrenocorticotropic hormone, and low testosterone and dehydroepiandrosterone sulfate, indicating congenital adrenal hyperplasia. Genetic analysis revealed a mutation in the CYP17A1 gene and a karyotype of 46, XY; hence, she was diagnosed with 17OHD.
Related collections
Most cited references11
- Record: found
- Abstract: found
- Article: not found
Congenital adrenal hyperplasia
- Record: found
- Abstract: not found
- Article: not found
17-hydroxylation deficiency in man.
- Record: found
- Abstract: found
- Article: not found