- Record: found
- Abstract: found
- Article: found
Roles of Growth Differentiation Factor 15 in Atherosclerosis and Coronary Artery Disease
review-article
21 August 2019
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Introduction
The majority of acute cardiovascular events (CVE) in patients are caused by occlusive
thrombosis because of rupture or erosion of atherosclerotic plaques.1 Growth differentiation
factor 15 (GDF‐15), a stress‐responsive member of the transforming growth factor‐β
(TGF‐β) cytokine superfamily, has been shown to be a strong and independent predictor
of mortality and disease progression in patients with atherosclerosis and coronary
artery disease (CAD), such as acute coronary syndromes (ACS) and stable angina pectoris.2
The development of atherosclerosis is dependent upon a high‐inflammatory content,
which has been shown to modulate lesion initiation, progression, and potentially devastating
thrombotic complications.3 Angiogenesis plays an important role in the progression
of atherosclerotic plaque and complications.4, 5, 6 Atherosclerosis and cancer arise
from multiple factors and are consolidated from the very early stages of development
up to the advanced forms in inflammatory processes. Uncontrolled cell proliferation
and oxidative stress and angiogenesis appear to be unifying causal factors in both
diseases.7 A local inflammatory state occurring in atherosclerotic lesions has been
implicated in angiogenesis through activation of endothelial cells, release of chemokines,
cytokines, growth factors, lipid mediators, proteases, and increase of endothelial
metabolic rate. The angiogenesis allows extravasation of the plasma component, leading
to future thromboembolic events.8, 9, 10, 11 GDF‐15 might be an acute phase modifier
of TGF‐βRII‐dependent proinflammatory responses to atherosclerotic plaque rupture
and thrombus formation12 (Figure). Although the exact biological functions of GDF‐15
are still poorly understood, it has been shown to regulate inflammatory and angiogenesis
pathways (Figure). GDF‐15 exhibits differing and even opposing functions under various
circumstances. For instance, GDF‐15 has proapoptosis, antiapoptosis, proangiogenesis,
antiangiogenesis, proinflammatory, and anti‐inflammatory properties.12, 13 Therefore,
GDF‐15 exhibits a complex pattern with beneficial and harmful functions. GDF‐15 promoter
contains p53‐transcription factor binding sites that are required and sufficient for
the induction of GDF‐15 expression.14 Activation of p53 is a fundamental cellular
response to inflammation, oxidative stress, hypoxia, telomere erosion, and oncogene
activation. The circulating levels of GDF‐15 reflect these acute and chronic inflammatory
conditions linked with atherosclerosis and CAD.
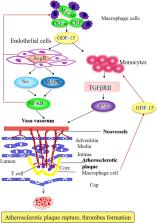
Figure 1
Schematic overview of a vulnerable plaque in advanced atherosclerosis. Plaque formation
is initiated by endothelial cell dysfunction and subsequent angiogenesis and release
of proinflammatory factors mediated by GDF‐15, contributing to the progression of
atherosclerotic lesions and the development of plaque rupture and thrombus formation
in atherosclerotic status. CRP indicates C‐reactive protein; GDF‐15, growth differentiation
factor 15; MAPK, mitogen‐activated protein kinase; M‐CSF, macrophage colony‐stimulating
factor; TGF‐βRII, transforming growth factor‐βRII; TNF‐α, tumor necrosis factor‐α.
Regulation and Roles of GDF‐15
Under normal physiological conditions, placenta is the only tissue expressing large
quantities of GDF‐15.15 GDF‐15 levels are increased in various pathological conditions
and diseases, including inflammation, cardiovascular disease, renal disease, pulmonary
disease, and cancer.12 GDF‐15 is produced in activated macrophages,11 and in pathological
conditions including proinflammatory status, vascular injury, pressure overload, and
oxidative stress from human endothelial cells,16 vascular smooth muscle cells,17 and
adipocytes.18 The expression of GDF‐15 in virtually all tissues suggests its importance
in general and basic cellular functions. Although the exact biological functions of
GDF‐15 remain largely unclear, it has been demonstrated to modulate inflammatory,
apoptotic, and angiogenesis pathways.
GDF‐15 as a Novel Biomarker of CVE
GDF‐15 has been recognized as a consistent biomarker of CVE in patients with ACS or
stable CAD.19 GDF‐15 levels are independently related to age, high‐sensitivity C‐reactive
protein (hs‐CRP), natriuretic peptides, and renal dysfunction in patients with established
CAD.2, 20, 21, 22, 23, 24, 25, 26 GDF‐15 concentrations are enhanced in patients with
multivessel disease21, 27 and with a history of myocardial infarction (MI) or heart
failure.27, 28, 29 The association of GDF‐15 with all‐cause mortality, cardiovascular
mortality, MI, and stroke was further explored in our recently published research
work, which included 3440 patients with established CAD independent of clinical predictors
including age, diabetes mellitus, current smoking, hypertension, hyperlipidemia, and
left ventricular ejection fraction.30 Our study simultaneously evaluated the incremental
prognostic value of GDF‐15 and provided more information than other biomarkers (estimated
glomerular filtration rate, fibrinogen, D‐dimer, sST2, pregnancy‐associated plasma
protein A, and uric acid). Adding the information on GDF‐15 to the baseline clinical
model improved the C‐index from 0.786 to 0.806. In addition, we examined whether there
were heterogeneity in the hazard ratios based on presentation with stable CAD and
ACS (unstable angina pectoris, non–ST‐segment–elevation myocardial infarction [NSTEMI],
and ST‐segment–elevation myocardial infarction [STEMI]) beyond traditional risk factors.
GDF‐15 was significantly associated with stable CAD and ACS. Recently, Gohar et al31
revealed that high circulating levels GDF‐15 are predictive of secondary CVE in women
with carotid atherosclerosis, indicating contribution of high GDF‐15 levels to increased
risk factors of CVE.
Roles of GDF‐15 in ACS
GDF‐15 is emerging as a prognostic biomarker in patients with ACS, including STEMI,
NSTEMI, and unstable angina pectoris (Table 1), which result from the rupture or erosion
of vulnerable atherosclerotic plaque leading to death and recurrent MI, which would
be occurring at any time after the first attack episode.32, 33, 34, 35, 36, 37, 38,
39, 40, 41 The predictive value of GDF‐15 has been confirmed in the 2 large non–ST‐segment–elevation
ACS (NSTE‐ACS) trials: the GUSTO‐IV (Global Utilisation of Strategies to Open Occluded
Arteries IV) and FRISC II (Fast Revascularization during Instability in Coronary Artery
Disease II) cohorts20, 21 (Table 1). As shown in patients from the GUSTO‐IV trial,
GDF‐15 concentrations are closely related to all‐cause mortality in NSTE‐ACS20 (Table 1).
In FRISC II, cumulative 1‐year mortality rates were 1.5, 5.0, and 14.1% in patients
with low, moderately increased, and markedly increased concentrations of GDF‐15. GDF‐15
provided prognostic information beyond clinical predictors and other prognostic biomarkers,
including cardiac troponin T, N‐terminal pro‐brain natriuretic peptide, hs‐CRP, and
creatinine clearance.21 The independent association of GDF‐15 with mortality is confirmed
in other patients with STEMI or NSTE‐ACS.22, 23 Lately, the prognostic value of GDF‐15
has been reevaluated in 16 876 patients with NSTE‐ACS or STEMI randomized to ticagrelor
or clopidogrel in the PLATO (Platelet Inhibition and Patient Outcomes) study27(Table 1).
Based on the large number of patients and outcome events, the PLATO biomarker study
was able to explore the relation of GDF‐15 to specific outcome events during follow‐up.
After adjustment for clinical predictors and other biomarkers, higher GDF‐15 concentrations
were associated with an increased risk of all‐cause mortality, cardiovascular mortality,
MI, and stroke. The results are confirmed by a secondary analysis of the PLATO study
including 17 095 patients with ACS,42 demonstrating that GDF‐15 was a strong marker
associated with all‐cause death, death caused by other vascular or nonvascular causes,
and death caused by bleeding (Table 1). For GDF‐15, the possible signal of association
with death caused by bleeding is in line with prior results indicating that GDF‐15
reflects nonoverlapping disease pathway contributing to the development of bleeding
after ACS. Increased concentrations of GDF‐15 also identify patients at increased
risk for adverse left ventricular remodeling and hospitalization for heart failure
after ACS.23, 28, 43 In 3501 patients from the PROVE IT–TIMI‐22 (Pravastatin or Atorvastatin
Evaluation and Infection Therapy‐Thrombolysis in Myocardial Infarction‐22) trial,
GDF‐15 was associated with the risks of all‐cause mortality, recurrent MI, and hospitalization
for new or worsening heart failure.28 The prognostic information provided by GDF‐15
was independent of clinical predictors and other biomarkers (hs‐CRP and brain natriuretic
peptide). Notably, GDF‐15, in contrast to hs‐CRP,44 did not decline over time in response
to more intensive statin therapy in PROVE IT–TIMI‐22,28 further indicating that GDF‐15
reflects a nonoverlapping atherosclerotic pathway contributing to the development
of ACS.
Table 1
GDF‐15 Related to Outcome Events in ACS
Study
Participants
Outcomes
Follow‐Up (y)
Comparisons (ng/L)
RR (95% CI)
CAD patients, Kempf et al2
ACS (n =877)
M
6 (maximum)
<1200, 1200 to 1800, >1800
8.5 (3.81–18.99)
GUSTO‐IV, Wollert et al20
NSTE‐ACS (n=2081)
M
1 (maximum)
<1200, 1200 to 1800, >1800
2.08 (1.85–2.34)
FRISC‐II, Wollert et al21
NSTE‐ACS (n=2079)
M, R
2 (maximum)
<1200, 1200 to 1800, >1800
1.75 (1.48–2.07)
ASSENT‐2 and ASSENT‐plus trials, Kempf et al22
STEMI (n =741)
M
1 (maximum)
<1200, 1200 to 1800, >1800
6.6 (2.43–18.23)
AMI patients, Khan et al23
AMI (n=1142)
M, HF
1.4 (median)
1470 (240–31 860)
4.24 (3.21–5.62)
FRISC II, Eggers et al24
NSTE‐ACS (n=950)
M, R
0.5 (maximum)
<1200, 1200 to 1800, >1800
1.9 (1.2–3.0)
PLATO, Hagstrom et al27
ACS (n=16 876)
M
1 (maximum)
<1145, 1145 to 1550, 1550 to 2219, >2219
3.96 (2.91–5.39)
PROVE IT‐TIMI 22, Bonaca et al28
ACS (n=3501)
M
2 (maximum)
<1200, 1200 to 1800, >1800
4.76 (2.67–8.48)
STEMI patients, Eitel I et al32
STEMI (n=238)
M,R
0.5 (maximum)
<1319, ≥1319
19 (2.58, 139.66)
NSTE‐ACS patients, Widera et al33
NSTE‐ACS (n=1122)
M, R
0.5 (mean)
1725 (1205–2797)
2.4 (1.9–3.0)
NSTE‐ACS patients, Widera et al34
NSTE‐ACS (n=1146)
M, R
0.5 (mean)
1770 (1262–2981)
2.4 (2.0–3.0)
ICTUS, Damman et al35
NSTE‐ACS
M
5 (maximum)
<1200, 1200 to 1800, >1800
4.78 (3.71–6.18)
PLATO, Wallentin et al36
NSTE‐ACS (n=9946)
M,R,S
1 (maximum)
<1200, 1200 to 1800, >1800
NA
NSTE‐ACS patients, Dominguez‐Rodriguez et al37
NSTE‐ACS (n=255)
M,R,UA
3 (maximum)
1639 (median)
52.3 (7–388.5)
Shock II, Fuernau et al38
AMI (n=190)
M
0.1 (maximum)
7662 (median)
1.88 (1.21–2.94)
FRISC‐II, Wallentin et al39
NSTE‐ACS (n=2457)
M,R
2 (maximum)
<1800, ≥1800
NA
NSTE‐ACS patients, Dominguez‐Rodriguez et al40
NSTE‐ACS (n=502)
M,R,UA
2 (maximum)
470 to 1765, 1766 to 2995, 2996 to 11 607
6.6 (4.28–10.2)
Västmanland Myocardial Infarction Study, Skau et al41
AMI (n=847)
M
6.9 (median)
NA
2.57 (2.31–2.85)
PLATO, Lindholm et al42
ACS (n=17 095)
M
1 (maximum)
NA
2.65 (2.17–3.24)
ACS indicates acute coronary syndrome; AMI, acute myocardial infarction; ASSENT, assessment
of the Safety and Efficacy of a New Thrombolytic; CAD, coronary artery disease; FRISC
II, Fast Revascularization during Instability in Coronary artery disease II; GDF‐15,
growth differentiation factor 15; GUSTO‐IV, Global Utilisation of Strategies to Open
Occluded Arteries IV; HF, heart failure; ICTUS, Invasive versus Conservative Treatment
in Unstable coronary Syndromes; M, mortality; NA, not applicable; NSTE‐ACS, non‐ST‐segment–elevation
acute coronary syndrome; PLATO, Platelet Inhibition and Patient Outcomes; PROVE IT‐TIMI‐22,
Pravastatin or Atorvastatin Evaluation and Infection Therapy‐Thrombolysis in Myocardial
Infarction‐22 trial; R, recurrent myocardial infarction; RR, relative risk; S, stroke;
STEMI, ST‐segment–elevation myocardial infarction; UA, unstable angina.
Roles of GDF‐15 in Stable CAD
GDF‐15 maintains its close association with an adverse prognosis in patients with
ACS during the transition to the chronic stage of CAD.24, 25 In a serial analysis
from FRISC‐II, GDF‐15 provided similar independent prognostic information on the composite
end point of death or recurrent MI on admission and up to 6 months after an episode
of NSTE‐ACS.24 Similarly, GDF‐15 was identified as an independent predictor of CAD
mortality in patients with stable CAD2 (Table 2). In the AtheroGene (patients with
stable CAD or ACS who had at least 1 stenosis >30% in a major coronary artery were
enrolled in the AtheroGene registry) study, which included 1352 patients with stable
angina pectoris undergoing coronary angiography, GDF‐15 was associated with CAD mortality
independent of cardiovascular risk factors, clinical predictors, the number of diseased
vessels, left ventricular ejection fraction, and other biomarkers (cTnI, N‐terminal
pro‐brain natriuretic peptide, and hs‐CRP).2 Similarly, in a cohort of 984 patients
with stable CAD, higher GDF‐15 levels were associated with lower left ventricular
ejection fraction, worse diastolic function, and greater inducible ischemia. The association
of GDF‐15 with MI, heart failure, and cardiovascular death persisted after extensive
adjustment for traditional risk factors and the other biomarkers (NT‐proBNP, CRP,
and hs‐cardiac troponin T)29 (Table 2). Recently, the prognostic value of GDF‐15 has
been reevaluated in 14 577 patients with stable CAD in specific outcome events from
STABILITY (The Stabilization of Atherosclerotic Plaque by Initiation of Darapladib
Therapy) study 26 (Table 2). Our recent study further validated that GDF‐15 is associated
with cardiovascular and noncardiovascular death (eg, cancer morbidity) in stable CAD
patients with and without previous cancer diagnosis.30 Furthermore, our study also
indicated the independent associations between the GDF‐15 and coronary thrombotic
events (eg, MI), even after adjusting for other prognostic biomarkers (estimated glomerular
filtration rate and left ventricular ejection fraction).
Table 2
GDF‐15 related to outcome events in stable CAD
Study
Participants
Outcomes
Follow‐Up (y)
Comparisons (ng/L)
RR (95% CI)
Kempf et al2
Stable CAD (n=1352)
M
3.6 (median)
1128 (850–1553)
2.7 (2.2–3.3)
Dallmeier et al25
Stable CAD (n=1029)
M
10 (median)
1232 (916–1674)
2.80 (1.98–3.37)
Hagstrom et al26
Stable CAD (n=14 577)
M
3.7 (median)
1253 (915–1827)
2.63 (1.91–3.63)
Schopfer et al29
Stable CAD (n=948)
M
8.9 (mean)
2166 (1589–3057)
2.97 (2.58–3.43)
CAD indicates coronary artery disease; GDF‐15, growth differentiation factor 15; M,
mortality; RR, relative risk.
GDF‐15 is a biomarker considered for introduction to the clinic. What questions remain
to be answered to establish GDF‐15 as a clinically useful biomarker? Moreover, is
GDF‐15 a risk biomarker or a causative risk factor, or more importantly, what are
the circumstances under which GDF‐15 is just a marker of risk versus a causative factor?
Its function as a protective or disease‐inducing factor remains largely unknown. The
GDF‐15 puzzle is a good example of how epidemiological and mechanistic studies can
interact successfully. The predictive value persists even a decade later, and the
findings discussed above support the hypothesis that GDF‐15 is not a consequence of
cardiovascular disease or a passive biomarker of the disease process, but in fact
plays an active role in the pathophysiology of atherosclerosis and CAD.45, 46 The
clinical significance of newly discovered mechanisms can be evaluated and conversely,
the mechanisms behind epidemiologically proven associations can be elucidated.
GDF‐15 and Inflammation in Atherosclerosis
Potential mechanisms have been suggested for the association of GDF‐15 with adverse
outcomes in atherosclerosis, including worse baseline cardiac disease severity, inflammation,
ischemia, volume overload, and adipokines.12 Elevated GDF‐15 has been shown to promote
inflammation and angiogenesis,47, 48, 49 implying that GDF‐15 may play an important
role in the pathogenesis of atherosclerosis. While GDF‐15 is a cardiovascular risk
factor, whether GDF‐15 contributes directly to atherosclerosis development has not
been established and the precise relationships between GDF‐15 and atherosclerosis
are incompletely understood. GDF‐15 was originally identified as a factor overexpressed
in activated macrophages to regulate inflammation, which is involved in all stages
of atherosclerosis, from its initiation and progression to its thrombotic complications.
de Jager et al49 demonstrate that leukocyte deficiency of GDF‐15 improves atherosclerotic
plaque stability by impairing macrophage migration and promoting collagen deposition.
GDF‐15 deficiency in leukocytes is associated with reduced macrophage accumulation
in an atherosclerosis model, suggesting a pro‐inflammatory role of GDF‐15 in atherosclerosis.
Moreover, chromatin immunoprecipitation assays confirmed that p53 was recruited to
both p53 binding sites 1 and 2 in the GDF‐15 promoter in response to CRP.50 Accordingly,
CRP induces GDF‐15 expression through the regulation of p53 binding sites in the GDF‐15
promoter. Along this line, GDF‐15 is involved in orchestrating atherosclerotic lesion
progression by regulating apoptotic cell death and IL‐6‐dependent inflammatory responses
to vascular injury.51 These data suggest an involvement of GDF‐15 in the initiation
and progression of atherosclerosis. GDF‐15 revealed a central role for this factor
as a pro‐inflammatory cytokine that accelerates atherosclerosis.
GDF‐15 is in fact associated with subclinical atherosclerosis.52 GDF‐15 deficiency
resulted in a reduction of early atherosclerotic lesion size after 4 weeks on a high
cholesterol Western‐type diet. After 12 weeks, no differences in lesion size could
be detected.49 It is known that lesions in mice become quite complex with increased
duration of feeding.53 Moreover, GDF‐15 expression is significantly higher in acute
stages of human plaque rupture (unstable angina pectoris) than in advanced stable
lesions (stable angina pectoris). Paradoxically, overexpression of GDF‐15 in macrophages
significantly attenuates atherosclerotic lesions in the ApoE−/− mouse model of atherosclerosis.54
GDF‐15 is thought to have anti‐inflammatory effects on cells, including cardiomyocytes.55
Preusch et al demonstrated a proinflammatory plaque phenotype in mice transplanted
with bone marrow from GDF‐15−/− donors with enhanced macrophage accumulation, suggesting
a protective effect of GDF‐15 on the atherosclerosis process.56 However, this effect
may contribute to changes in lesion vulnerability such as thinning of fibrous caps
and potential plaque rupture. It should, however, be noted that they did not focus
on the onset of atherosclerotic changes within the vascular wall such as lipid accumulation
in younger mice. It is known as a model of late‐stage disease in atherosclerosis and
does not show much progress in early stages. To further elaborate on this, de Jager
et al investigated the signal transduction cascades for GDF‐15. Blockade of TGFβRII,
but not TGFβRI/ALK5, abrogated the GDF‐15‐elicited MCP‐1 response, suggesting the
role of GDF‐15 in the underlying mechanism of atherosclerosis progress. Thus, GDF‐15
has a pleiotropic regulatory effect on the inflammatory process, in line with that
of other TGF‐β family members such as activin‐A57 and TGF‐β1.58 Previous study pointed
out that expression of GDF‐15 may be upregulated by a variety of proinflammatory stimuli
in macrophages including interleukin (IL)‐1β, IL‐2, and tumor necrosis factor‐α.11
Recent study found a positive association between the IL‐1β and CVE,59 suggesting
there is an interleukin‐1β/GDF‐15‐associated immunity pathway resulting in atherosclerosis.
Accordingly, the high levels of GDF‐15 may result from high levels of monokines such
as IL‐1β, tumor necrosis factor‐α, and CRP. GDF‐15 initiates pro‐ and anti‐inflammatory
effects on atherosclerosis development and progression, depending on the pathophysiological
context and progression stage. GDF‐15 functions as a proinflammatory factor in the
process of atherosclerosis via TGFβRII signaling, especially in the early stage and
acute inflammatory stage, leading to vulnerable plaque, which provides 1 of the possible
mechanisms for the atherosclerosis process.
GDF‐15 and Angiogenesis
Plaques that are most at risk are characterized by large necrotic cores with a thin
fibrous cap. Plaque angiogenesis and intraplaque hemorrhage are important contributors
to unstable lesions.6, 60 Although commonly regarded as separate disease entities,
there is a growing recognition that cardiovascular disease and cancer have various
similarities with shared common biology and risk factors, including age, diabetes
mellitus, hypertension, smoking, physical inactivity, and unhealthy diet. A novel
function for GDF‐15 was identified as a potent angiogenic factor to be secreted from
melanoma cells together with vascular endothelial growth factor to promote vascular
development.61 During angiogenesis, endothelial cells emerge from the quiescent state
and undergo progression in the cell cycle. GDF‐15 is causally involved in the pathological
process of endothelial proliferation and angiogenesis.9, 62 Jin et al revealed the
functional effect of GDF‐15 on the cell cycle progression of endothelial cells and
demonstrated that GDF‐15 upregulates expression of cyclins D1 and E in human umbilical
vein endothelial cells, leading to a rapid transition from G1 to S phase.9 GDF‐15
has been shown to promote cell viability, invasion, migration, and angiogenesis in
HepG2 cells48 and hypoxic human umbilical vein endothelial cells possibly through
inhibiting p53 signaling.62 Moreover, GDF‐15 induced the pro‐angiogenic effects through
the phosphorylation of Src and its downstream pathways of AKT, MAPK, and NF‐κB signaling,
implying regulatory roles of GDF‐15 in cell proliferation and angiogenesis in atherosclerosis.47
Intriguingly, proinflammatory factors such as IL‐1β, tumor necrosis factor‐α, and
CRP induce GDF‐15 expression in macrophage cells through the regulation of p53 binding
sites in the GDF‐15 promoter (Figure). GDF‐15 promoted macrophage chemotaxis in a
strictly CCR2‐ and TGF‐β type II receptors (TGFβRII)–dependent manner in early and
advanced atherosclerosis. Accordingly, the GDF‐15/TGFβRII/P53 and the GDF‐15/NF‐κB
pathways are the critical mechanisms involved in the angiogenesis and acute inflammation
in the unstable atherosclerotic plaque. The functional proatherogenic roles of GDF‐15
in lesion progression indicate that besides other TGF‐β superfamily members such as
TGF‐β1 and BMP, interference with GDF‐15 may be a useful novel strategy for therapeutic
intervention.63, 64
GDF‐15 and Stress in Atherosclerosis
GDF‐15 and brain natriuretic peptide are similarly induced by biomechanical stress
in isolated rat cardiomyocytes and in the murine heart. GDF‐15 is upregulated in response
to stressors including in macrophages exposed to oxidized low‐density lipoprotein
in atherosclerotic carotid arteries.65 Specific to atherosclerosis, GDF‐15 has shown
predictive abilities of CAD mortality and composite outcomes in stable CAD and ACS
in patients with prevalent cardiovascular risk factors.2, 21, 24, 28 Recent findings
support that GDF‐15 is associated with subclinical atherosclerosis as assessed by
maximal internal carotid artery intima‐media thickness as well as the presence of
carotid plaque. Whether GDF‐15 is a mediator of cardiovascular disease or upregulated
in response to cardiovascular injury remains unclear. After further adjusting CRP
and brain natriuretic peptide, the association of GDF‐15 with maximum internal carotid
artery intima‐media thickness and carotid plaque was more robust. This suggests that
GDF‐15 may reflect an orthogonal pathway associated with cardiovascular disease, the
mechanism of which remains unclear.
Roles of GDF‐15 in Cancer and Other Diseases
GDF‐15 is characterized by a wide tissue distribution pattern with high expression
in the prostate and placenta, heart, intestine, liver, kidney, pancreas, colon, lung,
brain, and skeletal muscle.66 It acts as a multifunctional cytokine by controlling
numerous physiological and pathological processes. Acting on the hypothalamus and
hindbrain, GDF‐15 is a key inducer of cancer‐related anorexia and weight loss.67 Moreover,
GDF‐15 plays an important role in the physiological regulation of energy intake and
expenditure, with a more pronounced effect in women than in men.68 Although several
studies suggest antitumoral activity, the protumoral effects of GDF‐15 appear to prevail.12,
47, 69
Like the other members of the TGFβ‐superfamily, GDF‐15 has opposite effects depending
on cellular context, disease stage, or microenvironment. GDF‐15 has both antitumorigenic
and protumoral properties. In fact, these apparently paradoxical data could be explained
by a dual role of GDF‐15 in cancer progression: inhibition of carcinogenesis in normal
tissue at early stages of tumor development and promotion of tumor at late stages
of the disease.70 GDF‐15 induces pleiotropic effects in cancer by modulating cancer
cell proliferation and chemoprotection but also the tumoral microenvironment (angiogenesis,
invasion and metastasis processes, and immunomodulation), as well as more unexpected
processes (cancer‐induced anorexia). GDF‐15 has been implicated in chronic disease,
such as rheumatoid arthritis, end‐stage renal failure, or diabetes mellitus.71, 72,
73 As for cancer or cardiovascular diseases, GDF‐15 plasma concentration was an independent
predictor of disease worsening and/or death. The biological processes that could explain
such a link are obscure and often not known. A recent study emphasizes the positive
effects of GDF‐15 on peripheral nerve regeneration.74 In this case, GDF‐15 seems to
reduce the number of regenerated axons but it increases the maturation of newly formed
ones. This leads to better recovery of sensorimotor function.
Potential Implications of GDF‐15 in Atherosclerosis
GDF‐15 functions as a direct participant in the atherosclerotic process. Plaque angiogenesis
is a physiological response to the increased oxygen demand in the plaque but has adverse
effects by facilitating intraplaque hemorrhage and influx of inflammatory mediators.
The angiogenesis inhibitor angiostatin reduces plaque angiogenesis, and the secondary
reduction of macrophages may have beneficial effects on plaque stability.75 GDF‐15
deficiency contributes to angiogenesis and improves atherosclerotic plaque stability
by impairing macrophage migration and promoting collagen deposition. A high level
of serum GDF‐15 is detected in human atherosclerotic lesions,49, 65 which are broadly
proportional to the disease burden. Thus, we speculate that GDF‐15 is located in arteriosclerotic
lesions or in circulation and promotes atherosclerotic plaque vulnerability by increasing
angiogenesis and inflammation. Intriguingly, GDF‐15 promotes indirect proinflammatory
effects in atherosclerosis49, 51 but mediates anti‐inflammatory effects in acute MI
by directly inhibiting myeloid cell recruitment.76 Although it is possible that GDF‐15
itself could be causative in the development of ACS,54 GDF‐15 has anti‐apoptotic and
antihypertrophic properties of in cardiomyocytes subjected to simulated ischemia/reperfusion
injury.77 Notably, the biological effects of GDF‐15 are context dependent and may
vary with the stage of the disease.34, 36, 55, 56, 76, 78, 79, 80 In line with these
investigations, GDF‐15 has been shown to be associated with subclinical atherosclerosis
involved in macrophage accumulation in atherosclerosis.52 Correspondingly, GDF‐15
is responsible for early‐stage atherosclerotic lesions. Inflammatory factors (IL‐1β
or CRP) secreted from macrophages induce GDF‐15 expression through the regulation
of p53 binding sites in the GDF‐15 promoter further activates its downstream NF‐κB
signaling, accelerating the progression of atherosclerosis in the early stage, and
promoting the formation of vulnerable plaque. GDF‐15 is also linked with endothelial
dysfunction and more advanced coronary atherosclerosis, suggesting the regulatory
roles of GDF‐15 in chronic myocardial and vascular damage in the late stage of the
atherosclerosis process.19 More basic research into the pathobiological features of
GDF‐15 is needed to explore the mechanism related to the risk of new atherosclerosis
and recurrent ischemic events after ACS.
Potential Implications for GDF‐15 in ACS and CAD
GDF‐15 appears to be a very consistent marker of adverse long‐term outcome in ACS.
However, may GDF‐15 be used to identify groups of patients who will or will not benefit
from various interventions or treatments? Are there any treatments for which monitoring
of GDF‐15 concentrations might be useful to guide the treatment (dose and/or duration)?
Several studies illustrate the potential of the marker to risk stratify unselected
contemporary patient populations treated outside clinical trials. In a recent investigation
that compared the incremental prognostic value of 9 biomarkers on top of the GRACE
(Global Registry of Acute Coronary Events) score in unselected patients with NSTE‐ACS,
GDF‐15 emerged as the most promising biomarker.34 Underscoring its potential to add
information to what is clinically available, GDF‐15 also added discriminatory information
to GRACE when hs‐cardiac troponin T was considered as an additional continuous variable.
In accordance with previous observations, we noted in our population‐based cohort
that addition of GDF‐15 to standard cardiovascular risk factors resulted in modest
but significant improvements in the C‐statistic (discrimination) as well as reclassification,
as measured by the integrated discrimination improvement and net reclassification
improvement for all‐cause death and cardiovascular death.30 In addition, GDF‐15 predicted
all‐cause mortality more accurately independently of hs‐cardiac troponin T and N‐terminal
pro‐brain natriuretic peptide in patients with acute pain.81 The PLATO trial showed
that GDF‐15 contributes information on the magnitude of benefit by a successful intervention
such as ticagrelor regardless of invasive or noninvasive management.36 The FRISC‐II
trial supported that a high concentration of the biomarker GDF‐15 implies heightened
risk and functions as a useful identification of patients who might expect the longest
postponement of death or MI with an early invasive strategy.39 Thresholds offer a
convenient way to classify patients into risk categories that may be linked to treatment
decisions. However, the use of thresholds may reduce statistical power given the continuous
association of GDF‐15 with cardiovascular risk. Alternatively, GDF‐15 might be incorporated
as a continuous variable into established or novel risk scores that can be presented
as nomograms or applications on (handheld) electronic devices. Therefore, in a setting
with a need for prioritization among different patients with NSTE‐ACS for early invasive
procedures, direct access to treatment for patients with elevated troponin, in addition
to a fast track for those with high GDF‐15, might be a useful strategy. New algorithms
for decision support in ACS are currently under evaluation, including variables such
as troponin and GDF‐15 showing significant interactions with the effects of an early
invasive treatment strategy. More importantly, there are medical therapies that reduce
the risk of cardiovascular disease and cancer. For example, use of daily aspirin for
the primary prevention of major CVE reduces the incidence of cancer and cancer mortality,
although more research is required to identify which individuals are likely to benefit
most.82 In previous studies of patients with NSTE‐ACS, GDF‐15 has been found to predict
future events and contribute to the identification of high‐risk patients with a benefit
of an early invasive strategy. Thus, GDF‐15 might be of clinical value in refining
risk stratification and tailoring treatment of patients with ACS. GDF‐15 provides
unique information on underlying disease processes leading to a raised risk of severe
events, eg, fatal CVE and death.27 Moreover, the previous studies published on the
functional proatherogenic role of GDF‐15 in lesion progression indicate that besides
other TGF‐β superfamily members such as TGF‐β1 and BMP,83 interference with GDF‐15
may be a useful novel strategy for therapeutic intervention.48, 51 Therefore, circulating
levels of GDF‐15 are suggested as a prognostic marker to improve risk stratification
of patients with ACS, along with benefit from treatment with a high‐dose, highly efficient
statin. Increased GDF‐15 plasma concentrations at the time of PCI and stent implantation
might classify high‐risk patients with ACS who benefit from high‐dose, highly efficient
statins, implicating that high‐dose statins are more effective in high‐risk patients
and obtaining GDF‐15 may help identify these patients.45, 46
Conclusions and Future Directions
GDF‐15 functions as a cardiovascular risk and outcome marker and appears to be a direct
participant in the atherosclerotic process. GDF‐15 is responsible for vulnerable atherosclerotic
lesions by proinflammation and angiogenesis, accelerating the progression of atherosclerosis
especially in the early stage, subsequently contributing to promotion of the vulnerable
plaque formation. Moreover, GDF‐15 has been found to predict CVE and identify high‐risk
patients with a benefit of an early invasive strategy. The importance of the GDF‐15/TGFβRII/P53
and the GDF‐15/NF‐κB signaling pathways in the cardiovascular system sparked hopes
that manipulating its pathophysiological activity could provide novel therapeutic
agents for atherosclerosis and CAD. In fact, the clinical and experimental studies
clearly support a physiological and pathophysiological role for the GDF‐15 system
in atherosclerosis and CAD. Targeting the GDF‐15 pathway represents a novel therapeutic
approach against atherosclerosis and CAD that will increase our understanding of the
pathophysiology of these diseases.
Sources of Funding
This research program was supported by the National Major Research Plan Training Program
of China (91849111) and the National Natural Science Foundation of China (81770253;
81670214; 81370362), Natural Cultivation Foundation of the Capital Medical University
(PYZ2018106) and Talent project of Beijing Chaoyang Hospital Affiliated to Capital
Medical University.
Disclosures
None.
Related collections
Most cited references75
- Record: found
- Abstract: found
- Article: not found
Transforming growth factor-beta regulation of immune responses.
Ming Li, Yisong Y Wan, Shomyseh Sanjabi … (2006)
- Record: found
- Abstract: found
- Article: not found
MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily.
M Bootcov, A Bauskin, S. M. Valenzuela … (1997)
- Record: found
- Abstract: found
- Article: not found
Tumor-induced anorexia and weight loss are mediated by the TGF-beta superfamily cytokine MIC-1.
Heiko Johnen, Shu Lin, Tamara Kuffner … (2007)