- Record: found
- Abstract: found
- Article: found
First Detection of an Enterovirus C99 in a Captive Chimpanzee with Acute Flaccid Paralysis, from the Tchimpounga Chimpanzee Rehabilitation Center, Republic of Congo
Read this article at
Abstract
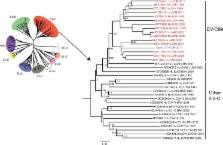
Enteroviruses, members of the Picornaviridae family, are ubiquitous viruses responsible for mild to severe infections in human populations around the world. In 2010 Pointe-Noire, Republic of Congo recorded an outbreak of acute flaccid paralysis (AFP) in the humans, caused by wild poliovirus type 1 (WPV1). One month later, in the Tchimpounga sanctuary near Pointe-Noire, a chimpanzee developed signs similar to AFP, with paralysis of the lower limbs. In the present work, we sought to identify the pathogen, including viral and bacterial agents, responsible for this illness. In order to identify the causative agent, we evaluated a fecal specimen by PCR and sequencing. A Human enterovirus C, specifically of the EV-C99 type was potentially responsible for the illness in this chimpanzee. To rule out other possible causative agents, we also investigated the bacteriome and the virome using next generation sequencing. The majority of bacterial reads obtained belonged to commensal bacteria (95%), and the mammalian virus reads matched mainly with viruses of the Picornaviridae family (99%), in which enteroviruses were the most abundant (99.6%). This study thus reports the first identification of a chimpanzee presenting AFP most likely caused by an enterovirus and demonstrates once again the cross-species transmission of a human pathogen to an ape.
Related collections
Most cited references35
- Record: found
- Abstract: found
- Article: not found
Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination.
- Record: found
- Abstract: found
- Article: not found
Molecular evolution of the human enteroviruses: correlation of serotype with VP1 sequence and application to picornavirus classification.
- Record: found
- Abstract: found
- Article: not found