
1. INTRODUCTION
The gut microbiome encodes many enzymes with crucial roles in the biotransformation of diverse xenobiotics and endogenous substances [1]. Among the reported gut-microbiota-produced enzymes in mammals, bile salt hydrolases (BSHs; E. C.3.5.1.24), a group of cysteine hydrolases, are essential in deconjugation of the glycine- or taurine-conjugated bile acids (BAs) [2, 3], via catalyzing hydrolytic reactions of the C24 amide bond of conjugated BAs into unconjugated BAs and free glycine or taurine. BAs are key endogenous substances in humans, which have many important biological functions, such as facilitating the digestion and absorption of dietary lipids, as well as acting as hormones or signaling molecules targeting distinct receptors [4–6]. Parts of BAs have been reported to be potent ligands for a range of host nuclear receptors (such as farnesoid X receptor and pregnane X receptor) [7] and G protein-coupled receptors [8], thereby strongly influencing host lipid and glucose metabolic and immunomodulatory processes [9, 10]. Accumulating evidence indicates that BSH abundance and activity are tightly associated with BAs homeostasis and a variety of human diseases including inflammatory bowel disease, obesity, type 2 diabetes, and liver and cardiovascular diseases [11–14], thus suggesting that BSHs are key targets for treating metabolic diseases.
Although multiple natural and synthetic compounds have been found to have BSH inhibitory activity in the past few decades, very few compounds have been reported to have potent inhibition and good safety profiles [15, 16]. Therefore, discovering more effective BSH inhibitors is urgently needed. Given that natural products remain the major source for the identification of drug leading compounds, we initiated large-scale screening to assess the inhibitory potential of natural products toward lsBSH. After preliminary assessment of six classes of natural products (more than 100 natural products), we identified several naturally occurring chalcones with strong to moderate lsBSH inhibition activity ( Figure 1 ). Chalcones are widely found in many medicinal plants and herbs [17, 18], which have been reported to have diverse biological activities, such as anti-inflammatory effects and inhibitory activity against enzymes including tyrosine kinase, aldose reductase, cyclooxygenase, carboxylesterases and pancreatic lipase [19–23]. However, the BSH inhibition activity of naturally occurring chalcones and their inhibitory mechanisms have not been reported and investigated.
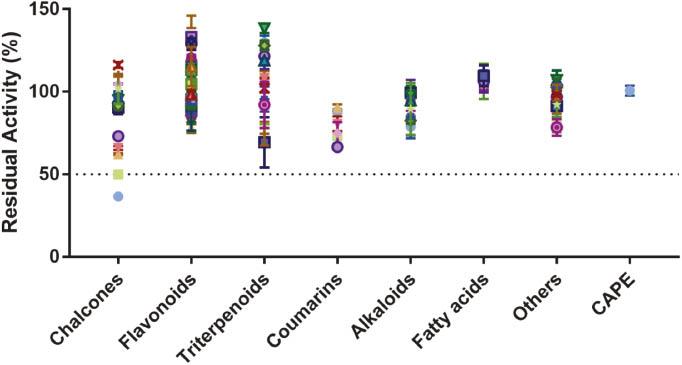
Inhibitory effects of more than 100 types of natural products (1 μM final concentration) on lsBSH.
All data are shown as mean ± SD of triplicate determinations.
The objectives of this study were to determine the inhibitory activity of naturally occurring chalcones on BSH in vitro and to investigate the inhibitory mechanisms of two newly identified potent inhibitors of lsBSH. To this end, a series of inhibition assays were first performed to assess the inhibitory potential of chalcones on lsBSH, and the inhibitory mechanisms of licochalcone C and isobavachalcone (two most potent inhibitors against lsBSH) were investigated in detail through a range of inhibition kinetic assays, fluorescence quenching assays and docking simulations. These findings should advance medicinal chemists’ understanding of the inhibitory mechanisms of chalcones against bacterial BSHs and support the development of more efficacious BSH inhibitors for modulating BA metabolism.
2. MATERIALS AND METHODS
2.1 Chemicals and reagents
Licochalcone A, licochalcone B, licochalcone C and licochalcone D were purchased from Vikeqi Biology Technology Co., Ltd. (Sichuan, China). Naringenin chalcone, flavokawain A, 4′-O-methylbroussochalcone B and taurocholic acid (TCA) were ordered from Yuanye Bio-Technology Co., Ltd. (Shanghai, China). Butein and dithiothreitol (DTT) were ordered from J&K Chemical Ltd. (Beijing, China). Caffeic acid phenethyl ester (CAPE) was ordered from Dalian Meilun Biotech Co., Ltd. (Dalian, China). Isoliquiritigenin, echinatin, isobavachalcone, bavachalcone, isoliquiritin, neoisoliquiritin and other natural compounds used in BSH inhibition assays were obtained from Chengdu Pufei De Biotech Co., Ltd. (Chengdu, China), and 2′,4′-Dihydroxy-6′-methoxy-3′,5′-dimethylchalcone was provided by Mr. Wei Xing of East China University of Science and Technology. The purity of all tested natural compounds exceeded 98%. Glycerol, imidazole, sodium acetate, NaCl, lysozyme and phenylmethylsulfonyl fluoride were ordered from Sinopharm Chemical Reagent Co., Ltd. SuperNuclease was obtained from Sino Biological Inc. The cholic acid-AMCA probe (CA-AMCA) was synthesized by author Peng-chao Huo, whereas its hydrolytic metabolite 7-amino-4-methyl-3-coumarinylacetic acid (AMCA) was provided by Energy Chemical Ltd. (Shanghai, China). LC grade methanol, acetonitrile, DMSO and formic acid were ordered from Tedia (Fairfield, OH, USA) and used carefully, and 100 mM phosphate buffered saline (PBS, pH 6.0) was prepared with ultrapure water.
2.2 Expression and purification of BSHs
Expression constructs for amino-terminally fused BSHs (lsBSH, btBSH or efBSH) in a pET29a (+) vector were overexpressed in Escherichia coli (E. coli) [14, 24, 25]. The expression plasmid was transformed into BL21 (DE3) cells, and expression was induced through the auto-induction method at 18°C. After 36 h of induction, cells were harvested by centrifugation and resuspended in 40 mL lysis buffer (25 mM Tris-HCl, pH 8.0, mixed with 150 mM NaCl, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride, 0.1 mg/mL lysozyme and 25 U/mL SuperNuclease). After 30 min at room temperature, the cells were lysed by sonication on ice and centrifuged at 18000 rpm at 4°C for 30 min. Subsequently, 2 mL Ni-NTA agarose was mixed with supernatant, and the lsBSHs was eluted with elution buffer (25 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM DTT and 100–200 mM imidazole). The eluted protein was purified on a Superdex 200 10/300 GL column equilibrated in protein storage buffer (10 mM sodium acetate, pH 5.5, 400 mM NaCl and 1 mM DTT) ( Figure S1 ).
2.3 Chemical synthesis of CA-AMCA
CA-AMCA was synthesized according to a published method ( Figure S2 ) [26]. Both 1H- and 13C-NMR spectra were used for structural characterization of CA-AMCA, which were acquired on a Bruker AVANCE instrument, with DMSO-d6 as the solvent. Chemical shifts were recorded by using tetramethylsilane as the reference. 1H NMR (600 MHz, DMSO-d6) d (ppm): 10.34 (s, 1H), 7.91–7.64 (m, 2H), 7.49 (dd, J = 8.8, 2.1 Hz, 1H), 4.33 (s, 1H), 4.13 (s, 1H), 4.06–3.95 (m, 1H), 3.80 (s, 1H), 3.62 (s, 1H), 3.53 (d, J = 18.0 Hz, 2H), 3.19 (dt, J = 11.0, 6.3 Hz, 1H), 2.41 (ddd, J = 14.7, 10.1, 4.6 Hz, 1H), 2.36 (s, 3H), 2.32–2.11 (m, 3H), 2.05–1.94 (m, 1H), 1.91 (s, 1H), 1.89–1.71 (m, 5H), 1.71–1.60 (m, 2H), 1.51–1.41 (m, 3H), 1.41–1.15 (m, 7H), 1.03–0.93 (m, 4H), 0.81 (s, 3H), 0.60 (s, 3H). 13C NMR (150 MHz, DMSO-d6) d (ppm): 173.02, 161.43, 152.76, 148.82, 142.53, 126.41, 115.61, 115.59, 105.64, 71.47, 70.90, 66.70, 46.55, 46.22, 41.98, 41.85, 40.52, 35.78, 35.62, 35.37, 34.86, 34.04, 31.70, 30.87, 29.05, 27.75, 26.69, 23.29, 23.10, 21.64, 17.64, 15.37, 12.83. ESI-MS m/z: 624.35 [M+H]+ ( Figures S3–S5 ).
2.4 LC-FD analysis of CA-AMCA and its hydrolytic product AMCA
Both CA-AMCA and AMCA were analyzed with an LC system equipped with a RF-20A fluorescence detector (Shimadzu, Kyoto, Japan) ( Figure S6 ). Chromatographic separation was performed on a Shim-pack VP-ODS C18 column (4.6 mm × 150 mm, 5 mm, Shimadzu, Japan), and the column temperature was kept at 40 ˚C. The fluorescence signals of AMCA and CA-AMCA were recorded with an excitation wavelength at 345 nm and emission wavelength at 455 nm. The mobile phase was a mixture of acetonitrile (A) and 0.2% formic acid water (B) with the following gradient conditions: 0–2 min, 75%–55% B; 2–3 min, 55%–20% B; 3–5 min, 20%–10% B; 5–5.1 min, 10%–75% B; and 5.1–7 min, maintenance at 75% B. The flow rate was 0.5 mL/min. The standard curve for AMCA and an enzymatic kinetic plot of lsBSH-catalyzed CA-AMCA hydrolysis are shown in Figures S7 and S8 , respectively.
2.5 BSHs inhibition assays using CA-AMCA as a substrate
Briefly, a mixture (190 μL) containing lsBSH (2 μg/mL final concentration), DTT (1 mM final concentration) and various inhibitors was pre-incubated in 100 mM PBS (pH 6.0) at 37°C for 3 min. Subsequently, 10 μL reaction buffer containing 0.2 mM CA-AMCA was added to start the biotransformation. The reaction was terminated with ice-cold acetonitrile (200 μL) after incubation at 37°C for 30 min. After centrifugation at 20000 × g for 30 min, 100 μL of the supernatant was transferred into an LC vial and analyzed with LC-FD. The residual activity of BSH was calculated as: (peak area of AMCA in the presence of inhibitor)/peak area of AMCA in negative control (DMSO only) × 100%.
2.6 Inhibition of TCA hydrolysis by licochalcone C and isobavachalcone
In brief, a mixture (190 μL) containing lsBSH (2 μg/mL), DTT (1 mM) and various inhibitors was pre-incubated in 100 mM PBS (pH 6.0) at 37°C for 3 min. Subsequently, 10 μL of PBS containing 1 mM TCA was added to start the biotransformation. The reaction was terminated with ice-cold acetonitrile (200 μL) containing the internal standard diclofenac after incubation at 37°C for 30 min. Then the mixtures were centrifuged at 20000 × g for 30 min, and 100 μL of the supernatant was diluted with 100 μL ultrapure water. The samples were analyzed with LC-MS/MS according to a published method with minor modifications [27].
2.7 Inhibition kinetic analyses of licochalcone C and isobavachalcone
To explore the inhibitory mechanisms of the two natural chalones against lsBSH, we performed a series of inhibition assays with various concentrations of the fluorescent substrate CA-AMCA and increasing concentrations of inhibitors. The inhibition kinetics was determined by the intersection point in the Lineweaver-Burk plot. The slopes of the lines in the Lineweaver-Burk plot were then used to depict the second plot and to determine the inhibition constant (Ki ) value [28–30]. The following equations for competitive (a), non-competitive (b) or mixed inhibition (c) were used.



where V is the velocity of CA-AMCA hydrolyzed by lsBSH, and Vmax is the maximum velocity. S and I are substrate (CA-AMCA) and inhibitor concentrations, respectively. Ki is the inhibition constant of the inhibitors toward lsBSH, and Km is the Michaelis constant for CA-AMCA hydrolyzed by lsBSH.
2.8 Fluorescence quenching assays
Fluorescence spectra of lsBSH with or without inhibitor were recorded with a Duetta fluorescence and absorption spectrometer (HORIBA Scientific). In brief, 1 mL of 100 mM PBS (pH 6) containing lsBSH (20 μg/mL), TCEP (0.25 mM) and inhibitors in increasing concentrations (0, 0.25, 0.5, 1, 1.5, 2, 3 and 4 μM) was used. After incubation at 25 ˚C for 10 min, the mixture was transferred to a cuvette and scanned at an excitation wavelength of 274 nm. The fluorescence quenching mechanism was described by the Stern-Volmer equation:

where F0 and F are the fluorescence intensity in the absence and presence of inhibitors, respectively. KSV is the Stern-Volmer dynamic quenching constant. Kq is the bimolecular fluorescence quenching rate constant. τ0 is the fluorescence lifetime of the fluorescent group without the quencher, and [Q] is the concentration of inhibitors. For biological macromolecules, the average value of τ0 was approximately 10−8 s.
2.9 Molecular docking simulations
Docking simulations were performed with AutoDock Vina (Version 1.1.2). First, the crystal structure of lsBSH (PDB ID: 5YP7) was obtained from Protein Data Bank (https://www.rcsb.org/). Then the hydrogens were added, and the Kollman charges were assigned. The search space was set to 70 × 70 × 70 °C Ǻ3, centered on the catalytic residue Cys2 with a spacing of 0.375 Ǻ. The docking modes with the lowest binding energy were displayed and further analyzed.
2.10 Inhibition of BSHs in mouse feces by licochalcone C and isobavachalcone
Fecal pellets collected form six healthy ICR mice were suspended in PBS and then broken into fine particles to a final concentration of 50 mg/mL. The mixtures were centrifuged at 9000 × g for 30 min at 4°C, and the supernatants were mixed in equal volumes. The reaction solutions (100 μL) contained the fecal solution (1 mg/mL, final concentration), DTT (1 mM, final concentration), inhibitors in increasing concentrations and CA-AMCA (10 μM, final concentration). The reaction was quenched with ice-cold acetonitrile (300 μL) after incubation at 37°C for 30 min. After centrifugation at 20000 × g for 30 min, 100 μL of the supernatant was transferred into an LC vial and analyzed by LC-FD.
3. RESULTS AND DISCUSSION
3.1 Screening of lsBSH inhibitors from a natural-product library
To identify strong BSH inhibitors from natural products, we measured the inhibitory potential of 110 natural compounds against lsBSH at three inhibitor concentrations (1 μM, 10 μM and 100 μM). As presented in Figure 1 and Table S1, several naturally occurring chalcones strongly inhibited lsBSH, and licochalcone C and isobavachalcone showed the greatest inhibition, with more than 50% activity loss at a dose of 1 μM. In contrast, the residual activity of lsBSH in the presence of the other tested compounds, including the positive lsBSH inhibitor (CAPE), exceeded 50% at a dose of 1 μM. This finding encouraged us to further study the inhibitory effects of naturally occurring chalcones against lsBSH and other bacterial BSHs.
3.2 Inhibition on lsBSH-catalyzed CA-AMCA hydrolysis by chalones
Among all tested chalcones, two chalcone glycosides (isoliquiritin and neoisoliquiritin) and flavokawain A (a polymethoxylated chalcone) showed very weak lsBSH inhibitory activity even at a high inhibitor concentration (100 μM). To quantify the inhibitory potency of another 12 natural chalcones, we plotted the dose-response curves with increasing inhibitor concentrations. As shown in Figure 2 and Figure S9 , all tested chalcones dose-dependently inhibited lsBSH-catalyzed CA-AMCA hydrolysis. The chemical structures and IC50 values of all tested chalcones are listed in Table 1 . Of these, licochalcone C and isobavachalcone showed the most effective lsBSH inhibitory activity, with IC50 values less than 1 μM. Meanwhile, the inhibitory effects of bavachalcone, 4′-O-methylbroussochalcone B, licochalcone D, licochalcone A and 2′,4′-dihydroxy-6′-methoxy-3′,5′-dimethylchalcone were also stronger than that of the positive inhibitor CAPE (IC50 = 13.64 μM, Figure S10 ). Other tested chalcones displayed moderate inhibition, with IC50 values ranging from 10 μM to 100 μM. These results clearly suggested that parts of naturally occurring chalcones potently inhibited lsBSH, some of which are more potent than previously reported positive lsBSH inhibitors.

Dose-inhibition curves of six natural chalcones (IC50 < 5 μM) against lsBSH-catalyzed CA-AMCA hydrolysis.
(A) Licochalcone C, (B) isobavachalcone, (C) bavachalcone, (D) 4′-O-methylbroussochalcone B, (E) licochalcone D, (F) licochalcone A. All data are shown as mean ± SD of triplicate determinations.
Chemical structures of chalcones and their IC50 values against lsBSH. All data are shown as mean ± SD of triplicate determinations
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Name | C2′ | C3′ | C4′ | C5′ | C6′ | C2 | C3 | C4 | C5 | IC50 (μM) | |
| 1 | Isoliquiritigenin | –OH | –H | –OH | –H | –H | –H | –H | –OH | –H | 14.90 |
| 2 | Echinatin | –H | –H | –OH | –H | –H | –OMe | –H | –OH | –H | 33.17 |
| 3 | Naringenin chalcone | –OH | –H | –OH | –H | –OH | –H | –H | –OH | –H | 22.42 |
| 4 | Butein | –OH | –H | –OH | –H | –H | –H | –OH | –OH | –H | 17.23 |
| 5 | Flavokawain A | –OH | –H | –OMe | –H | –OMe | –H | –H | –OMe | –H | >100 |
| 6 | Isobavachalcone | –OH |
 | –OH | –H | –H | –H | –H | –OH | –H | 0.92 |
| 7 | Bavachalcone | –OH | –H | –OH |
| –H | –H | –H | –OH | –H | 1.15 |
| 8 | 4′-O-Methylbroussochalcone B | –OH | –H | –OMe |
| –H | –H | –H | –OH | –H | 1.23 |
| 9 | Licochalcone B | –H | –H | –OH | –H | –H | –OMe | –OH | –OH | –H | 74.42 |
| 10 | Licochalcone A | –H | –H | –OH | –H | –H | –OMe | –H | –OH |
 | 3.73 |
| 11 | Licochalcone C | –H | –H | –OH | –H | –H | –OMe |
| –OH | –H | 0.78 |
| 12 | 2′,4′-Dihydroxy-6′-methoxy-3′,5′-dimethylchalcone | –OH | –CH3 | –OH | –CH3 | –OMe | –H | –H | –H | –H | 5.34 |
| 13 | Licochalcone D | –H |
| –OH | –H | –H | –OMe | –OH | –OH | –H | 1.73 |
| 14 | Isoliquiritin | –OH | –H | –OH | –H | –H | –H | –H | –Glu | –H | >100 |
| 15 | Neoisoliquiritin | –OH | –H | –Glu | –H | –H | –H | –H | –OH | –H | >100 |
After carefully analyzing the inhibitory effect of these natural chalcones on lsBSH, we summarized the structural-inhibition relationships of chalcones as lsBSH inhibitors. First, the addition of a glucosyl group on the chalcone skeleton was found not to be beneficial for lsBSH inhibition. In contrast, the chalones with multiple hydroxyl groups showed good lsBSH inhibition. When the hydroxyl groups at C-4′, C-6′ and C-4 were replaced by methoxy groups, the inhibition effect was weakened, as evidenced by naringenin chalcone (IC50 = 22.42 μM) vs flavokawain A (IC50 > 100 μM). Moreover, the introduction of an unsaturated alkyl chain in the A or B ring was beneficial for lsBSH inhibition, as evidenced by isobavachalcone (IC50 = 0.92 μM), bavachalcone (IC50 = 1.15 μM) vs isoliquiritigenin (IC50 = 14.90 μM), as well as licochalcone C (IC50 = 0.78 μM), Licochalcone A (IC50 = 3.73 μM) vs echinatin (IC50 = 33.17 μM). These findings should be very useful for the medicinal chemists in designing and developing more efficacious chalcone-type lsBSH inhibitors.
3.3 Inhibition of lsBSH-catalyzed TCA hydrolysis by licochalcone C and isobavachalcone
Given that conjugated BAs are the physiological substrates of lsBSH, we investigated the inhibitory effects of two chalcones on lsBSH-catalyzed TCA hydrolysis. As shown in Figure S11 , both licochalcone C and isobavachalcone strongly inhibited lsBSH in a dose-dependent manner, with IC50 values of 2.57 μM and 5.84 μM, respectively. These results clearly showed that licochalcone C and isobavachalcone inhibited the hydrolysis of both the optical substrates and physiological substrates catalyzed by lsBSH; therefore, these agents may modulate BA metabolism in the gastrointestinal system.
3.4 Inhibitory mechanisms of licochalcone C and isobavachalcone against lsBSH
Next, a series of inhibition kinetic analyses were performed to explore the inhibitory mechanisms of licochalcone C and isobavachalcone against lsBSH-catalyzed CA-AMCA hydrolysis. First, time-dependent inhibition assays indicated that the inhibition tendency and IC50 values of licochalcone C and isobavachalcone were not time dependent, thus suggesting that these two agents were reversible inhibitors of lsBSH ( Figure S12 ). We then determined the inhibition modes and the inhibition constants (Ki ) of these two natural compounds against lsBSH. As shown in Figure 3 , the Lineweaver-Burk plots of licochalcone C and isobavachalcone against lsBSH-catalyzed CA-AMCA hydrolysis showed that these two compounds strongly inhibited lsBSH via mixed inhibition, with Ki values of 0.73 μM and 1.00 μM, for licochalcone C and isobavachalcone, respectively. These findings clearly demonstrated that both licochalcone C and isobavachalcone effectively and reversibly inhibit lsBSH-catalyzed hydrolytic reactions.

3.5 Fluorescence spectroscopy analysis
Fluorescence quenching assays were also performed to study the interactions of licochalcone C and isobavachalcone with lsBSH. As depicted in Figure 4A and B , the maximum inherent fluorescence emission of lsBSH was observed around 322 nm, whereas the fluorescence intensity of lsBSH significantly decreased with the addition of increasing concentrations of licochalcone C or isobavachalcone. The quenching mechanism was further analyzed according to the Sterne-Volmer plot. As shown in Figure 4C and D , a good linear relationship between the F0/F and the concentration of inhibitors was obtained, thus suggesting that both licochalcone C and isobavachalcone quench the intrinsic fluorescence of lsBSH through a single quenching mechanism. The Ksv values of licochalcone C and isobavachalcone were calculated as 0.95 × 106 and 0.82 × 106 L/mol, respectively, whereas the Kq values were calculated as 0.95 × 1014 and 0.82 × 1014 L/mol/s, respectively. The Kq values of these two agents were much greater than the maximum scatter collision quenching constant of each type of enzyme quencher (2.0 × 1010 L/mol/s) [31]. These data indicated that licochalcone C and isobavachalcone quench the intrinsic fluorescence of lsBSH through a static mechanism caused by the formation of ligand-protein complexes, thereby affecting the microenvironment of some important fluorescent groups (such as Trp and Tyr) in lsBSH surrounding the ligand-binding sites.

3.6 Molecular docking simulations
To further investigate the potential ligand-binding sites and the interaction modes between the two newly identified lsBSH inhibitors and the target enzyme, we performed molecular docking simulations of either licochalcone C or isobavachalcone with the reported 3D structure of lsBSH (PDB ID: 5Y7P). Before docking, the potential ligand-binding pockets of lsBSH were predicted with CavityPlus, and the top two potential sites were selected on the basis of ligandability and druggability scores ( Figure S13 ). One was located at the catalytic site, which was relatively conserved and contained the key nucleophilic residue Cys2, whereas another was located below the back side of the catalytic site. As shown in Figure 5 , both licochalcone C and isobavachalcone docked well in the catalytic site or the back site. The spatial conformations of licochalcone C and isobavachalcone superimposed at the catalytic site or the back site are shown in Figure 5A . Consequently, the predicted binding energies of both licochalcone C and isobavachalcone on the catalytic site were calculated as -9.3 kcal/mol and -9.4 kcal/mol, respectively, and those on the back site were -6.7 kcal/mol and -7.0 kcal/mol, respectively, thus implying that both agents tightly bound the catalytic site of lsBSH.

Stereodiagram of licochalcone C and isobavachalcone superimposed at the catalytic site and the back site (A). The detailed view shows that licochalcone C (green) and isobavachalcone (orange) dock well into the catalytic site (B, C) and back site (D, E) of lsBSH.
The catalytic residue of lsBSH (Cys2) is shown as yellow sticks, and the key residues of lsBSH are shown as blue sticks.
At the catalytic site of lsBSH, the benzene rings of the two chalcones formed strong p-p interactions with Phe65 and Phe100, respectively. In addition, licochalcone C strongly interacted with Gly77 and Leu134 via hydrogen bonding, whereas isobavachalcone formed a hydrogen bond with Phe130 of lsBSH ( Figure 5B and C , Figure S14 ). In the back site, two benzene rings of the chalcones formed strong hydrophobic interactions with Leu182 and Phe230, respectively. Meanwhile, a hydrogen bonding interaction between isobavachalcone and Tyr-185 was additionally observed ( Figure 5D and E , Figure S15 ). These findings clearly indicated that licochalcone C and isobavachalcone stably bind lsBSH at two different ligand-binding sites, thus explaining the mixed inhibition modes of these two agents against lsBSH-catalyzed CA-AMCA hydrolysis. In addition, the interaction of these two agents with Phe and Tyr at each ligand-binding site partly explained the fluorescence quenching effects on the endogenous fluorescence of lsBSH, as shown in the results of the fluorescence quenching assays.
3.7 Inhibition of licochalcone C and isobavachalcone on other bacterial BSHs
Next, the inhibitory effects of licochalcone C and isobavachalcone on other bacterial BSHs (including btBSH and efBSH) were tested. As shown in Figure 6 , both licochalcone C and isobavachalcone inhibited btBSH and efBSH-catalyzed CA-AMCA hydrolysis in a dose-dependent manner. In btBSH, the IC50 values of licochalcone C and isobavachalcone were 7.46 μM and 5.17 μM, respectively. In efBSH, the IC50 values of these two agents were 14.01 μM and 7.91 μM, respectively. These results demonstrate that these two naturally occurring chalcones inhibit three various bacterial BSHs, thus suggesting that some chalcones can serve as broad-spectrum BSH inhibitors.

3.8 Inhibitory effects of licochalcone C and isobavachalcone against BSHs in mouse feces
Finally, the inhibitory effects of licochalcone C and isobavachalcone on the total activity of bacterial BSHs in mouse feces were evaluated. As shown in Figure 7 , both licochalcone C and isobavachalcone blocked the total activity of bacterial BSHs in mouse feces in a dose-dependent manner. The apparent IC50 values of these two compounds on the total activity of bacterial BSHs in mouse feces were determined to be 44.66 μM and 21.24 μM, for licochalcone C and isobavachalcone, respectively.

Dose-inhibition curves of licochalcone C (A) and isobavachalcone (B) against fecal BSH-catalyzed CA-AMCA hydrolysis.
All data are shown as mean ± SD of triplicate determinations.
As an important class of signal molecules in the mammals, BAs play a critical role in regulating host metabolism, including energy consumption, glucose and lipid homeostasis, as well as innate and adaptive immune responses [4, 32–34]. BSHs catalyze the critical first step in the metabolism of BAs by microorganisms in the intestinal tract, and their activity significantly influences BA characteristics and many physiological processes in the host [11, 35]. In recent studies, theabrownin from Pu-erh tea has been found to decrease serum and liver cholesterol levels through a mechanism associated with suppression of BSH related microorganisms and BSH activity [36]. In addition, berberine inhibits the activity of BSH in the intestinal flora and significantly increases the level of tauro-cholic acid in the intestines, thereby activating the intestinal FXR pathway and decreasing serum lipid levels [37]. Despite these important implications of BSHs activity, the lack of small-molecule inhibitors has limited deeper study, thus inspiring us to find efficacious BSHs inhibitors from natural products [38].
Chalcones, the intermediates in the biosynthesis of flavonoids, are widely found in medicinal plants such as Glycyrrhiza inflata Bat., Psoralea corylifolia L. and Carthamus inctorius L., and substantially contribute to the medicinal value of these herbs [39–42]. Moreover, these herbs have been shown to modulate BA metabolism [43–45]. In this study, natural chalcones were found to inhibit BSHs strongly or moderately, and these components readily contact BSHs after oral administration of the herbs. Given the low oral bioavailability and rapid hepatic metabolism of natural chalcones, these herbal medicines are highly likely to regulate BA metabolism partly by inhibiting BSHs in intestinal bacteria. Additionally, as a simple scaffold, chalcones can be easily synthesized through several routes, and various of chalcone derivatives have been synthesized and shown enhanced enzyme inhibition, bioavailability or in vivo efficacy [19, 46]. Together, these data and findings suggest that medicinal chemists can design and develop more powerful and innovative BSHs inhibitors by using chalcones as a scaffold, aiming to obtain ideal drug candidates for modulating BA metabolism and for treating BA-associated diseases.
4. CONCLUSION
In summary, this study screened the inhibitory potential of more than 100 natural compounds against lsBSH (a key gut-microbiota-produced cysteine hydrolase in deconjugation of conjugated BAs) and indicated that parts of naturally occurring chalcones were strong lsBSH inhibitors. Of all tested compounds, licochalcone C and isobavachalcone showed the most potent inhibitory effects against lsBSH. Further investigation demonstrated that these two naturally occurring chalcones strongly inhibited the lsBSH-catalyzed hydrolytic reaction in a mixed inhibition manner, with Ki values of 0.73 μM and 1.00 μM, respectively. Fluorescence quenching assays suggested that both licochalcone C and isobavachalcone quenched the intrinsic fluorescence of lsBSH through a static mechanism caused by the formation of stable ligand-protein complexes. Molecular docking simulations indicated that both licochalcone C and isobavachalcone stably bind lsBSH at two distinct ligand-binding sites, mainly via the formation of hydrogen bonds and hydrophobic interactions. Subsequent studies revealed that these two agents inhibited various bacterial BSHs and the total bacterial BSH activity in mouse feces. These findings demonstrate that licochalcone C and isobavachalcone are strong broad-spectrum BSH inhibitors and suggest that chalcones may be used as lead compounds for developing more efficacious BSH inhibitors.
