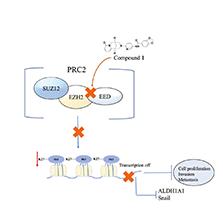
1. INTRODUCTION
Triple-negative breast cancer (TNBC) tumor metastasis is strongly associated with cancer stem cells (CSC) and the epithelial-mesenchymal transition (EMT) [1]. EMT activation confers cells with CSC characteristics, thus increasing their metastatic potential [2]. Compared with non-TNBC tissues, TNBC tissues have aberrant expression of CSC biomarkers (including CD133, CD44, CD24, and ALDH1A1), which are responsible for cancer progression and metastasis [3]. The classical EMT biomarkers in TNBC include E-cadherin, Snail, and Twist1, which regulate the mesenchymal cell phenotype in the dynamic process of EMT [4]. However, CSCs are often resistant to conventional chemotherapy, thus making TNBC particularly prone to metastasis and recurrence [5].
Embryonic ectoderm development (EED) interacts with EZH2 (enhancer of zeste homolog 2), thereby forming the essential catalytic subunit of the macromolecule polycomb repressive complex (PRC2) complex [6]. The EED-EZH2 protein-protein interaction (PPI) is essential for the methyltransferase activity of PRC2 [7]. Methylation of Lys27 of histone H3 (H3K27me3) leads to chromatin compaction and gene silencing [8]. Importantly, EED-EZH2 is highly expressed in TNBC and other solid tumors, where it activates the properties of CSC, increases metastasis, and promotes EMT [9–11]. The expression of EMT and CSC biomarkers has been associated with the activity of EED-EZH2 [12].
In recent years, EED-EZH2 PPI inhibitors have attracted considerable attention in cancer research [13]. A recent study has demonstrated that disrupting the EED-EZH2 PPI destabilizes PRC2 in cancer cells [14]. The US Food and Drug Administration (FDA)-approved EGFR inhibitor AZD9291 (osimertinib) has been reported to inhibit the EED-EZH2 PPI by targeting EZH2, thus resulting in tumor growth inhibition [15, 16]. However, strategies for disrupting the EED-EZH2 complex by targeting EZH2 have been hindered by single amino acid mutations at Y641 in EZH2, limited selectivity, and dose limitations [17–19]. In contrast, targeting the less highly conserved regions of the EED protein is a promising alternative strategy to disrupt the EED-EZH2 complex and inhibit PRC2 activity [20]. In recent years, several EED-EZH2 inhibitors that target EED have been developed. For instance, the FDA-approved drug astemizole inhibits cancer cell growth by targeting EED and blocking EED-EZH2 PPI, thereby arresting the proliferation of lymphomas [13]. Wedelolactone is a natural product compound that inhibits the proliferation of HepG2, THP1, and K562 cells in PRC2-dependent cancers by targeting EED [21]. Apomorphine hydrochloride was discovered as a potential EED-EZH2 interaction inhibitor that binds EED, and is used primarily in the treatment of Parkinson’s disease [22]. DC-PRC2in-01 inhibits cell proliferation and arrests the cell cycle in PRC2-driven lymphoma cells by targeting the EED-EZH2 interaction via EED [23]. However, to our knowledge, no EED-EZH2 PPI inhibitor has been reported for TNBC, particularly TNBC metastasis.
Herein, the cytisine analog compound 1 was identified as an EED-EZH2 PPI inhibitor that targets EED. Compound 1 directly blocked the interaction between EED and EZH2, thereby decreasing H3K27me3. Additionally, compound 1 potently inhibited CSCs and the EMT, and decreased 3D tumor sphere growth in vitro. Moreover, compound 1 selectively inhibited proliferation, and decreased migration and invasion in TNBC cells. This study demonstrates that targeting EMT and CSC signatures underlying the EED-EZH2 pathway may be a promising therapy for overcoming TNBC metastasis.
2. MATERIALS AND METHODS
2.1 Molecular modelling
The initial model of EED was derived from the X-ray crystal structure of EED with the inhibitor A-395 (PDB: 5K0M) through the molecular-conversion procedure implemented in the ICM-pro 3.6-1d program (Molsoft, San Diego, CA, USA) [24]. The molecular-conversion procedure and high-throughput molecular docking were performed as described in previous reports [25].
2.2 Materials and cell lines
Human TNBC cell lines (MDA-MB-231, BT549, and MDA-MB-468), non-TNBC cell lines (MCF-10A and T47D), human embryonic kidney (HEK 293T) cells, and normal liver (LO2) cells and were cultured Dulbecco’s modified Eagle’s medium supplemented with 1% penicillin and streptomycin, and 10% fetal bovine serum (Gibco). All cells were maintained at 37°C with 5% CO2 in a humidified atmosphere. Compounds 1–20 (purity > 95%) were purchased from J&K Scientific Ltd (Hong Kong, China). The positive control SAH-EZH2 (21) was purchased from Sigma-Aldrich. All compounds were dissolved in dimethyl sulfoxide (DMSO). Human EED protein was purchased from EpiGentek (Farmingdale, NY, USA). MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was obtained from Sigma-Aldrich (St. Louis, MO, USA).
2.3 Fluorescence polarization (FP) binding assay
The inhibitory effects of compounds on EED-EZH2 PPI were evaluated with FP assays as previously described [22, 26]. Briefly, compounds (1 μM in 1% DMSO) were transferred to black 384-well assay plates, and 625 nM His-EED and 20 nM FITC-labeled EZH2 (40–63) peptide tracer in FP buffer were then added and incubated at room temperature for 2 h. The negative control was 1% DMSO, and the positive control was 10 μM unlabeled EZH2 (40–63) peptide. Assay plates were read with a SpectraMax M5 microplate reader (Molecular Devices, San Jose, CA, USA) at 480 nm excitation and 535 nm emission with the FP measurement setting.
2.4 EED-knockdown assays
MDA-MB-231 cells were seeded in a flask and incubated until reaching 80% confluence. Lipo3000 reagent and EED siRNAs 5’-ATGGAGGATGATATAGATAAA-3 and 5’-CAGGCCATTTATTTCCCAGAA-3 were mixed with Dulbecco’s modified Eagle’s medium for 20 min at 37°C, then added to cells. At 48 h post-transfection, the cell density was 95% in the control group.
2.5 Cellular thermal shift assays
A total of 1 × 105 MDA-MB-231 cells were seeded in a 75 cm2 flask and incubated until 90% confluence was reached. Cell lysates were then collected and heated at various temperatures after incubation with compound 1 or DMSO for 30 min. The stabilizing effect of compound 1 was evaluated with western blotting.
2.6 Plasmid transfection
The pCMVHA EED plasmid (Addgene, 24231), Snail1 plasmid (YouBao, 40782), and ALDH1A1 plasmid (YouBao, 43362) were mixed with TurboFect reagent and used to transfect MDA-MB-231 cells in 25 cm2 flasks. Cells were incubated for 48 h before further use.
2.7 RT-qPCR assays
The inhibitory effects of compound 1 on ALDH1A1 and Snail were evaluated with RT-qPCR as previously described [27]. The primers were as follows. ALDH1A1 forward primer: GCACGCCAGACTTACCTGTC; ALDH1A1 reverse primer: CCTCCTCAGTTGCAGGATTAAAG; Snail forward primer: CCCAGTGCCTCGACCACTAT; Snail reverse primer: GCTGGAAGGTAAACTCTGGATTAGA; β-actin forward primer: AGGCACCAGGGCGTGAT; β-actin reverse primer: CTCTTGCTCTGGGCCTCGT.
2.8 Cell Counting Kit-8 (CCK-8) assays
Cells (MDA-MB-231, BT549, T47D, MCF-10A, LO2, and HEK 293T) were seeded in 96-well plates. After treatment with compound 1 for 72 h, 10 μL of CCK-8 solution buffer was added to the cells, which were incubated for 4 h. Finally, the optical density at 450 nm (OD 450) was detected according to the manufacturer’s instructions (Beyotime, C0038).
2.9 Lactate dehydrogenase (LDH) assays
Cells (MDA-MB-231, BT549, T47D, MCF-10A, LO2, and HEK 293T) were seeded in 96-well plates. After treatment with compound 1 for 72 h, cells were centrifuged at 400 g for 5 min, and the supernatant was removed. Then 150 μL of LDH buffer was added, and the cells were incubated for 1 h. After centrifugation at 400 g for 5 min, 120 μL/well supernatant was carefully transferred into a new 96-well plate. Finally, the OD 490 was detected according to the manufacturer’s instructions (Beyotime, C0016).
2.10 Colony-formation assays
The anti-proliferative activity of compound 1 against MDA-MB-231, BT549, T47D, and MCF-10A cells was evaluated with colony-formation assays as previously described [28]. Briefly, cells were seeded into six-well plates at a density of 1 × 104/well, then treated with 5 μM of compounds 1 or 21, or DMSO control, for 2 weeks. Colonies were stained with crystal violet and counted.
2.11 Chromatin immunoprecipitation (ChIP) assays
ChIP assays were performed according to a previous report with slight modifications [29, 30]. Briefly, anti-IgG (Cell Signaling Technology) and anti-EZH2 IgG (Santa Cruz Biotechnology) were used to capture DNA fragments after formaldehyde cross-linking. The samples were purified with a ChIP DNA Purification Kit (Active Motif, Carlsbad, CA, USA). The PCR primers for the target promoters are shown in Supplementary Table S1 .
2.12 Co-immunoprecipitation (co-IP) assays
Co-IP assays were performed according to a previous reported with slight modifications [31]. Cells were incubated with treatments with 5 μM of compounds 1 or 21, or DMSO, for 12 h. Cell lysates were collected after treatment and incubated with 10 μL pre-incubated anti-EZH2 magnetic beads, according to the manufacturer’s protocol. Finally, protein levels of EED were analyzed with western blotting.
3. RESULTS
3.1 Compound 1 is a potent and selective EED-EZH2 PPI inhibitor
The high-resolution X-ray structure of human EED in complex with A-395, an inhibitor of PRC2 that targets the H3K27me3-binding site of EED (PDB: 5K0M) [24], was used for virtual screening with the internal coordinate mechanics (ICM) method [ICM-Pro 3.6-1d program (Molsoft, San Diego, CA, USA)]. A natural product/natural product-like database of 90,000 compounds was docked to EED at the H3K27me3-binding site to identify potential inhibitors of EED. Twenty natural products ( Figure 1 ) with diverse structures exhibiting docking scores lower than −30.0 were shortlisted for biological testing with in vitro FP assays to measure disruption of the EED-EZH2 PPI ( Table 1 ). The clinical inhibitor SAH-EZH2 (21), which inhibits the EED-EZH2 PPI through targeting the EZH2-binding site on EED, was used as a positive control. Twelve hit compounds (1–5, 12–15, and 19–20) showed greater than 50% inhibition of the interaction between EED and EZH2 at 1 μM ( Figure 2A ). For reference, compound 21 disrupted the EED-EZH2 PPI by 51.2% at the same concentration. Compounds 1–3 and 12 also showed high cellular activity (described below) and were subjected to dose-response assays, which revealed half-maximal inhibitory concentration (IC50) values of 1.0 ± 0.6 μM, 2.8 ± 0.5 μM, 6.3 ± 1.9 μM, and 3.2 ± 1.4 μM against the EED-EZH2 PPI, respectively ( Figure 2B–2E ).

Structures of compounds 1–20, identified through high-throughput virtual screening, and positive-control compound 21.
ZINC numbers and docking scores of compounds tested in this study
| Name | ZINC no. | Relative molecular weight (Mr) | Scores |
|---|---|---|---|
| 1 | ZINC12662879 | 430.164 | −35.41 |
| 2 | ZINC02160442 | 481.200 | −34.22 |
| 3 | ZINC02096787 | 416.126 | −34.10 |
| 4 | ZINC04044926 | 399.158 | −32.63 |
| 5 | ZINC06174297 | 421.179 | −34.20 |
| 6 | ZINC02098018 | 456.157 | −35.40 |
| 7 | ZINC15967746 | 496.092 | −32.42 |
| 8 | ZINC08764329 | 431.137 | −40.59 |
| 9 | ZINC08877031 | 478.246 | −38.83 |
| 10 | ZINC96115184 | 448.189 | −35.15 |
| 11 | ZINC96115208 | 410.159 | −34.20 |
| 12 | ZINC96115443 | 489.119 | −37.17 |
| 13 | ZINC72324444 | 402.158 | −32.41 |
| 14 | ZINC12662879 | 508.176 | −38.31 |
| 15 | ZINC20412313 | 455.210 | −36.23 |
| 16 | ZINC12296841 | 441.169 | −34.87 |
| 17 | ZINC96221805 | 457.186 | −43.26 |
| 18 | ZINC96116572 | 375.170 | −40.54 |
| 19 | ZINC98363699 | 502.162 | −35.20 |
| 20 | ZINC96221432 | 437.195 | −35.43 |

Identification of compound 1 as the most potent EED-EZH2 PP1 inhibitor.
(A) Effects of compounds 1–21 on EED-EZH2 PPI, evaluated by FP assays. The structure of compound 1 is shown in the inset. (B-E) Dose-dependent inhibitory effects of compounds 1, 2, 3, and 12 on EED-EZH2 PPI, evaluated with FP assays. (F) Effect of compounds 1–5, 12–15, and 19–21 on ALDH1A1 mRNA levels in MDA-MB-231 cells, revealed by RT-qPCR. (G) Effects of compounds 1, 2, 3, and 12 on the expression of proteins downstream of H3K27me3 and ALDH1A1 in MDA-MB-231 cells, determined by western blotting.
The CSC biomarker ALDH1A1 has reported to be directly regulated by EZH2 [28]. The 12 hit compounds from the FP assays (1–5, 12–15, and 19–21) were further evaluated for their ability to decrease ALDH1A1 transcription in MDA-MB-231 TNBC cells ( Figure 2F ). Compounds 1–3 and 12 showed higher potency than the positive-control compound 21 in inhibiting ALDH1A1 expression. Moreover, western blotting was performed to evaluate the effects of compounds 1–3 and 12 in inhibiting H3K27me3 and ALDH1A1 protein expression in MDA-MB-231 cells. Compound 1 showed the greatest inhibition of H3K27me3 and ALDH1A1 protein expression, without any measurable effect on EED or EZH2 levels ( Figure 2G ). Finally, time-dependent western blotting indicated that the optimal inhibition activity was achieved at 12 h ( Supplementary Figure S1 ).
Compound 1 is a cytisine-based derivative, a compound class frequently used in drug design [32, 33]. Cytisine derivatives have shown promising biological activity for the treatment of various diseases, such as nicotine and alcohol dependency, cancers, and neurodegenerative diseases [34]. A cytisine-based inhibitor has been found to exhibit potent anti-proliferation effects against breast cancer cells through suppressing H3K4me3 and p27 expression [35, 36]. However, no biological activity of compound 1 itself had been reported.
3.2 Binding mode of compound 1 targeting EED-EZH2
In the PRC2 complex, EED has a regulatory role involving sensing the methylation status of H3K27me3-tagged histones, thus enabling positive allosteric control of EZH2 catalysis [37]. Specifically, engagement of the aromatic cage and top surface of EED by H3K27me3 stimulates the folding of an unstructured region of EZH2 into an alpha helix. This helix in turn stabilizes the binding site of the EZH2 SET domain [38]. Therefore, we hypothesized that molecules targeting the H3K27me3 site of EED might disrupt the EED-EZH2 interaction via an allosteric mechanism.
Molecular modeling was conducted to further elucidate the binding mode of compound 1 with EED. Molecular docking information for compound 1 in complex with EED indicated that compound 1 fits snugly within the H3K27me3-binding site on EED ( Figure 3A ). The cytisine core of compound 1 considerably overlaps with the binding pose of A-395 ( Figure 3B ); however the benzodioxole-containing side chain of compound 1 extends into an adjacent pocket that is not engaged by A-395. In this side pocket, one oxygen atom in the benzodioxole group forms a hydrogen bonding interaction with Trp364 on EED, a residue with an important role in maintaining the interaction of EED with histone H3 [39]. Because compound 1 was demonstrated to inhibit the EED-EZH2 PPI in vitro in FP assays, we hypothesized that the binding of compound 1 to the H3K27me3 site on EED might cause a conformational change in EED that prevents its interaction with EZH2.

Low-energy binding model for compound 1, generated by virtual screening. EED is displayed in ribbon form.
(A) Compound 1 and (B) A-395 are depicted as a ball-and-stick model showing carbon (yellow), hydrogen (grey), and oxygen (red) atoms. Hydrogen bonds are indicated as blue lines. The binding pocket of compound 1 is represented as a translucent green surface.
3.3 Compound 1 selectively binds EED and disrupts EED-EZH2 PPI
EZH2/1, SUZ12, and EED are the core subunits of the PRC2 complex. EZH1 shares 86% amino acid sequence identity with EZH2 [14]. SUZ12 is required to maintain the active conformation of the PRC2 complex [40]. To explore whether compound 1 directly binds EED over EZH2, EZH1, and SUZ12, we performed cellular thermal shift assays (CETSA). Lysates of MDA-MB-231 cells were treated with compound 1 (10 μM) or DMSO for 30 min, and CETSA analysis was then performed ( Figure 4A ). EED was significantly stabilized by compound 1 (ΔTm: 5.1 °C for EED), as compared with the DMSO control, thus suggesting that compound 1 directly engages EED even in the cellular environment ( Figure 4B ). Moreover, compound 1 exhibited no stabilizing effect on EZH2, SUZ12, and EZH1 ( Figure 4C–4E ); therefore, compound 1 appears to specifically target EED within the PRC2 complex. β-actin was used as a loading control and was not stabilized by 1 ( Figure 4F ).

Compound 1 selectively binds EED, thereby disrupting the EED-EZH2 PPI.
(A) CETSA analysis of compound 1 with EED, EZH2, EZH1, SUZ12, and β-actin. Cell lysates treated with 10.0 μM of compound 1, EED, EZH2, EZH1, SUZ12, and β-actin content in the soluble fraction, analyzed by western blotting. (B-F) Densitometry analysis of EED, EZH2, EZH1, SUZ12, and β-actin content. Data are represented as mean ± SD. *P < 0.05 vs DMSO group (Student’s t test).
3.4 Compound 1 decreases proliferation of MDA-MB-231 cells
The disruption of the EED-EZH2 complex leads to the inhibition of PRC2-driven cancers [13]. To study whether the anti-proliferative effect of compound 1 correlated with EED-EZH2 expression, we assessed its cytotoxicity in a panel of multiple cell lines. MDA-MB-231 and BT549 are human TNBC cell lines expressing high levels of EED-EZH2, whereas the human normal liver cell line (LO2), the human normal embryonic kidney cell line (HEK 293T), the human normal breast epithelial cell line (MCF-10A), and the human breast cancer cell line (T47D) express normal levels of EED-EZH2 ( Figure 5A ). We evaluated the anti-proliferation effects of compound 1 on HEK 293T, LO2, MCF 10A, T47D, MDA-MB-231, and BT549 cell lines with CCK-8 and LDH assays ( Figure 5B and 5C ). Compound 1 only slightly decreased the cell viability of all detected cells, according to CCK8 assays (IC50 > 100 μM) ( Figure 5B ), whereas it showed anti-proliferation activity toward MDA-MB-231 (IC50 = 30 ± 12.6 μM), BT549 (IC50 = 40 ± 9.8 μM), and T47D (IC50 = 80 ± 11.3 μM) cells in LDH assays. Differences in compound 1-mediated cytotoxicity of breast cancer cells were observed because LDH assays detect only cells with broken cell membranes, whereas CCK-8 assays detect all cells with intact mitochondria. Colony-formation assays further confirmed that compound 1 selectively inhibited the proliferation of MDA-MB-231 and BT549 TNBC cells ( Figure 5D ). Together, these results suggested that the anti-proliferative effects of compound 1 are associated with EED-EZH2 expression status.

The anti-proliferative effect of compound 1.
(A) EED-EZH2 protein levels in MDA-MB-231, BT549, T47D, MCF-10A, HEK 293T, and LO2 cell lines. (B-C) The viability of MDA-MB-231, BT549, T47D, MCF-10A, HEK 293T, and LO2 cell lines after treatment with different concentrations of compound 1 for 72 h, determined with CCK-8 and LDH assays. (D) Compound 1 shows selective anti-proliferation activity against MDA-MB-231 and BT549 cells in colonies. Data are represented as mean ± SD. **P < 0.01, ***P < 0.001 vs DMSO group (Student’s t test).
3.5 Compound 1 reverses the EMT and impairs CSC properties by targeting EED-EZH2 PPI
Because compound 1 exhibited promising EED-EZH2 PPI inhibitory activity, we explored its mechanism by using various biochemical assays. The co-IP and western blotting results suggested that compound 1 disrupts the interaction between EED and EZH2 without affecting the protein levels of EED and EZH2 in MDA-MB-231 cells ( Figure 6A and 6B ). Next, we investigated the effects of compound 1 on CSC and EMT biomarkers regulated by EED-EZH2, including Snail and ALDH1A1. Snail directly interacts with EED-EZH2 and subsequently represses the expression of E-cadherin during EMT [41]. Additionally, ALDH1A1 has recently been reported as a novel target of EZH2 [28]. EZH2 directly regulates ALDH1A1 expression in breast cancer cells, thus leading to CSC accumulation [42]. Hence, by directly regulating Snail and ALDH1A1, the EED-EZH2 complex promotes cell metastasis and invasion [28, 43]. In ChIP assays, cell lysates of compound 1-treated MDA-MB-231 cells were harvested and subjected to cross-linking and immunoprecipitation with anti-EZH2 and anti-IgG antibodies. Compound 1 suppressed the accumulation of the EED-EZH2 complex at ALDH1A1 and Snail promoters ( Figure 6C and 6D ). In agreement with this result, RT-qPCR revealed decreases in ALDH1A1 and Snail at the transcriptional level (primers in Supplementary Table S1 ) ( Figure 6E and 6F ). Moreover, compound 1 suppressed the protein levels of CSC biomarkers (ALDH1A1, CD44, and CD133) and an EMT biomarker (Snail), but promoted the expression of E-cadherin, a negative regulator of EMT ( Figure 6G ). Together, these results suggested that compound 1 may reverse the EMT and suppress the CSC biomarkers via blocking the EED-EZH2 PPI, thereby repressing the expression levels of its downstream genes.

Compound 1 reverses the EMT and CSC progress by inhibiting EED-EZH2 PPI.
(A-B) Effects of compound 1 on the EED-EZH2 PPI, evaluated with co-IP assays. (C-D) Compound 1 inhibits binding of EZH2 to the promoters of ALDH1A1 and Snail in MDA-MB-231 cells. ChIP assays were performed with primary antibodies against EZH2 and IgG. (E-F) Effects of compound 1 on the transcriptional levels of ALDH1A1 and Snail in MDA-MB-231 cells, determined by RT-qPCR. (G) Compound 1 inhibits CSC and EMT biomarkers in MDA-MB-231 cells by targeting the EED-EZH2 PPI. Data are represented as mean ± SD. *P < 0.05, **P < 0.01 vs DMSO group (Student’s t-test).
3.6 Compound 1 inhibits EMT and CSC protein levels in an EED-dependent manner
To further verify that compound 1 reverses the EMT and suppresses CSC biomarkers through targeting EED, we performed overexpression experiments. MDA-MB-231 cells were transfected with pCMWHA EED plasmid to induce EED overexpression. As shown in Figure 7A , compound 1 treatment significantly increased the protein levels of H3K27me3 and Snail, and decreased the expression of E-cadherin and ALDH1A1 in both EED-overexpressing and control MDA-MB-231 cells. However, the biological effects of compound 1 were more pronounced in the EED-overexpressing cells ( Figure 7B–7F ). Additionally, EED-knockdown cells were prepared through transfection of MDA-MB-231 cells with EED siRNA. EED-knockdown cells had lower levels of Snail and ALDH1A1, and higher expression of E-cadherin, than control cells. Importantly, knockdown cells were more resistant than control cells to compound 1 treatment, in terms of the decrease in Snail, ALDH1A1, CD44, CD133, and H3K27me3, and the increase in E-cadherin. Together, the results suggested that compound 1 targets EED-EZH2, thereby inhibiting EMT and CSC biomarkers.

Compound 1 regulates EMT and CSC biomarkers by targeting EED-EZH2.
(A) pCMWHA EED plasmid treatment induces EED overexpression in MDA-MB-231 cells. Western blotting of EZH2, EED, Snail, E-cadherin, ALDH1A1, H3K27me3, and β-actin control is shown. (B-F) Quantification of EED, E-cadherin, H3K27me3, Snail, and ALDH1A1 by western blotting. Data are represented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 vs DMSO group (Student’s t test).
3.7 The anti-metastasis effects of 1 are mediated by EED-EZH2 PPI
Three-dimensional tumor cell culture models are used to model human solid tumors in vitro in anticancer compound development [44]. The growth of tumor cells can induce the expression of proteins associated with metastasis and invasion [45]. Therefore, we evaluated the effects of compound 1 on the growth of 3D TNBC tumor spheroids ( Figure 8A ). The results indicated that compound 1 inhibits 3D tumor growth in a dose-dependent manner with higher potency than 21 in MDA-MB-231 cells ( Figure 8B ).
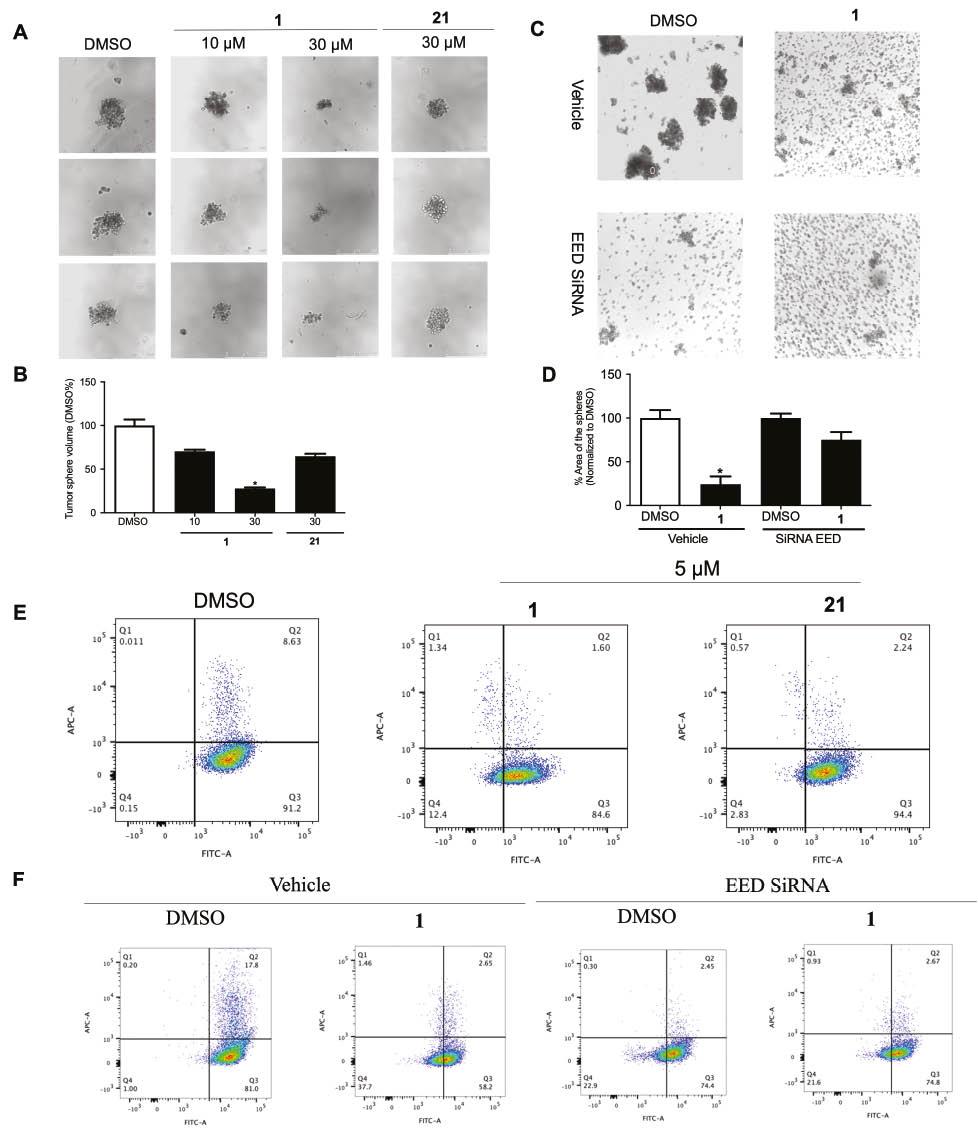
Compound 1 inhibits stemness in MDA-MB-231 cells.
(A) Compound 1 suppresses 3D tumor sphere growth in MDA-MB-231 cells. (B) Relative tumor sphere volume after treatment with compound 1. (C) Compound 1 inhibits 3D tumor sphere formation by blocking the EED-EZH2 PPI. (D) Relative tumor sphere volume. (E) Effects of compounds 1 and 21 (5 μM) on CD44+/CD24- populations in MDA-MB-231 cells, determined by flow cytometric analysis. (F) EED knockdown decreases CD44+/CD24- populations in MDA-MB-231 cells, and compound 1 has no further effect. Data are represented as mean ± SD. *P < 0.05 vs DMSO group (Student’s t test).
To further explore the role of the EED-EZH2 interaction in 3D tumor spheroid formation, we performed EED knockdown in MDA-MB-231 spheroids, which mimicked the effects of compound 1 treatment ( Figure 8C ). Importantly, in the EED-knockdown group, compound 1, compared with DMSO, induced no significant inhibition of 3D spheroid growth ( Figure 8D ).
The CD44+/CD24- population, a CSC biomarker, is high in TNBC [46]. The subpopulation of CD44+/CD24- cells, which has tumor-initiating capability in human breast tissue, is associated with breast cancer relapse and metastasis [47]. In flow cytometry experiments, compound 1 induced lower CD44+/CD24- populations in MDA-MB-231 cells than did DMSO or compound 21 ( Figure 8E ). Additionally, EED knockdown mimicked the effects of compound 1 treatment in decreasing CD44+/CD24-, and compound 1 induced no further effects in EED-knockdown cells ( Figure 8F ). Together, these results suggested that compound 1 exerts its anti-metastatic effects through inhibiting the EED-EZH2 PPI.
3.8 Compound 1 decreases migration and invasion of in MDA-MB-231 cells in an EED-EZH2-dependent manner
The activation of EMT and CSC promotes migration ability and metastasis [48, 49]. To confirm the role of EED-EZH2 inhibition against metastasis in TNBC, we evaluated the anti-migration and anti-invasion effects of compound 1 in MDA-MB-231 cells. EED-overexpressing MDA-MB-231 cells showed higher migration and invasion activity than control MDA-MB-231 cells. Compound 1 treatment inhibited migration and invasion in both EED-overexpressing and control cells. Notably, EED overexpression enhanced the anti-migration and anti-invasion effects of compound 1 ( Figure 9A–9C ). Additionally, to further explore whether EED is required for migration and invasion in TNBC, we performed EED knockdown in MDA-MB-231 cells. EED-knockdown cells showed lower cell migration and invasion, and were more resistant to the anti-metastasis activity of compound 1 ( Supplementary Figure 3 ). Together, these results suggested that the anti-metastasis effect of compound 1 on TNBC cells is associated with its ability to target the EED-EZH2 PPI. Snail and ALDH1A1 are genes downstream of the EED-EZH2 pathway that regulate metastasis. ALDH1A1 and Snail transfection partially rescued MDA-MB-231 cells from compound 1-induced anti-metastatic activity ( Figure 9D–9I ), thus suggesting that that compound 1 exerts its anti-metastatic effects through affecting these genes downstream of EED-EZH2.

Compound 1 inhibits migration and invasion of MDA-MB-231 cells by directly targeting EED.
(A) Migration and invasion ability of MDA-MB-231 cells transfected with vehicle or pCMVHA EED and treated with compound 1. (B-C) Quantitative analysis of cell migration and invasion. (D-I) Migration and invasion ability of MDA-MB-231 cells transfected with vehicle, FLAG ALDH1A1 plasmid, or FLAG SNAIL1 plasmid, and treated with compound 1. Data are represented as mean ± SD. *P < 0.05, **P < 0.01 vs DMSO group (Student’s t test).
4. DISCUSSION
EZH2 plays an important role in cancer development [50]. High levels of EZH2 are associated with aggressiveness in various cancer types and lead to poorer outcomes [50–52]. Therefore, EZH2 has attracted attention as an anti-cancer target. EZH2 inhibitors have been under pre-clinical and clinical investigation. However, many EZH2 inhibitors have shown therapeutic effects against only some hematological malignancies, and they may also cause drug resistance [53]. The enzymatic activity of EZH2 is regulated by EED in the PRC2 complex [26]. Therefore, targeting the interaction between EZH2 and EED is an alternative strategy against cancer. To our knowledge, no reports have studied the use of an EED-EZH2 PPI inhibitor to inhibit TNBC metastasis.
Natural or natural-like products have provided diverse scaffolds for drug development [54, 55]. In this study, a potent EED-EZH2 small-molecule inhibitor, compound 1, was developed through virtual screening of the ZINC natural product library. Although cytisine compounds have been reported against breast cancer, no biological activity of compound 1 has been described in the literature. CETSA analysis confirmed that compound 1 selectively engages EED in the cellular environment, thus disrupting the EED-EZH2 PPI in cellulo.
The EED-EZH2 PPI directly regulates CSC properties and promotes metastasis in cancer development [55]. In addition, PRC2 promotes EMT in metastasis through regulation of EMT biomarkers such as Snail [41, 56]. Using various biological assays, we showed that compound 1 exhibited more potent inhibitory effects on EMT and CSC biomarkers than the clinical EED-EZH2 PPI inhibitor compound 21. Mechanistically, compound 1 decreased the accumulation of EZH2 at the Snail and ALDH1A1 promoters, thereby abrogating Snail and accumulation of E-cadherin at the transcriptional and translational levels. Experiments using EED knockdown and overexpression suggested that compound 1 regulated EMT and CSC biomarkers by targeting the EED-EZH2 PPI. CSC and EMT signatures are consistently observed in TNBC cells, and these factors directly enhance metastasis [57]. Compound 1 inhibited the growth of 3D tumor spheroids, and also exhibited the anti-migration and anti-invasion ability of TNBC cells in an EED-dependent manner. Our experiments therefore confirmed the role of the EED-EZH2 interaction in promoting EMT and CSC signatures that drive the proliferation and metastasis activity of TNBC cells.
5. CONCLUSIONS
We developed the first reported cytisine-based EED-EZH2 PPI inhibitor against TNBC metastasis through structure-based virtual screening. Compound 1 selectively binds EED, thereby disrupting the EED-EZH2 PPI, altering EMT and CSC signatures, and decreasing metastasis in TNBC cells. Our data provide evidence that the cytisine scaffold may serve as drug candidates for the development of more potent and selective small-molecule inhibitors against EED-EZH2 overexpressing cancers.
