
Abbreviations: HNSCC, head and neck squamous cell carcinoma; NPC, nasopharyngeal carcinoma; TCM, traditional Chinese medicine; EMT, epithelial-mesenchymal transition; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor kappa-B; PI3K, phosphatidylinositol 3-kinase; AKT, protein kinase B; mTOR, mammalian target of rapamycin; JAK, Janus tyrosine kinase; STAT, signal transducers and activators of transcription; Nrf2, nuclear factor erythroid-2 related factor 2; CDKs, cyclin dependent kinases; MMP, mitochondrial membrane permeability; Bcl-2, B-cell lymphoma-2 protein; Bax, Bcl-2-associated X protein; Bak, Bcl-2 homologous antagonist/killer; Cyt C, cytochrome C; Apaf1, apoptosis protease activating factor 1; Fas-L, Fas ligand; TNF, tumor necrosis factor; ATG, autophagy-related protein; LC3 II, microtubule-associated protein 1 light chain 3 II; HCC, hepatocellular carcinoma; MMP-9, matrix metalloproteinase-9; FGF2, fibroblast growth factor-2; VEGF, vascular endothelial growth factor; SKP1, S phase kinase-associated protein 1; ECM1, extracellular matrix protein 1; ROS, reactive oxygen species; ATR, ataxia telangiectasia and rad3 related protein; CHK1, checkpoint kinase 1; CDDP, cisplatin; TUNEL, TdT-mediated dUTP nick-end labeling; ERK1/2, extracellular regulated protein kinases 1/2; HO-1, heme oxygenase-1; HIF-1α, hypoxia inducible factor-1α; PHDs, prolyl hydroxylases; AKR1C1, aldoketo reductase family 1 member C1; ARE, anti-oxidative response element; TKIs, tyrosine kinase inhibitors; RhoA, Ras homolog family member A; ROCK1, Rho-associated coiled-coil containing protein kinase 1; S6K, ribosomal protein S6 kinase; 4EBP1, eukaryotic translation initiation factor 4E binding protein 1; JNK, c-Jun N-terminal kinase; UVA, ultraviolet A; MAPKK, MAPK kinase; MAPKKK, MAPKK kinase; ERK5, extracellular regulated protein kinases 5; IκB, inhibitor of NF-κB protein; GSH, glutathione; NSCLC, non-small cell lung cancer; NQO1, NAD(P)H quinone dehydrogenase 1; γ-GCS, γ-glutamylcysteine synthetase; GCLC, glutamate-cysteine ligase catalytic subunit; GCLM, glutamate-cysteine ligase modifier subunit; PFOS, perfluorooctane sulphonate; PD-1, programmed death-1; BTK, Bruton’s tyrosine kinase; CDK4/6, cyclin dependent kinase 4/6; ALK, anaplastic lymphoma kinase; CSF3R, colony stimulating factor 3 receptors.
1. INTRODUCTION
Tumors are a consequence of uncontrolled proliferation that is associated with cell-cycle disruption and also involves inflammatory responses, apoptotic dysregulation, immune evasion and metastasis [1, 2]. Although a battle against cancer has been waged for centuries, it remains a major cause of death worldwide. The World Health Organization has reported that millions of people have a confirmed diagnosis of cancer every year [3–6]. Traditionally, four methods are used alone or in combination to combat cancer: surgery, radiotherapy, chemotherapy and immunotherapy [7]. Currently, for patients with advanced cancer, chemotherapy and radiotherapy are the leading therapy options, but they are limited by their severe adverse effects and the development of tumor resistance to chemotherapeutic drugs [8, 9]. Therefore, for better management of this mortal disease, novel treatments with minimal adverse effects, high accessibility and low cost are urgently needed.
Currently, traditional medicinal herbs, plants or their fruits containing beneficial bioactive constituents remain used by much of the world’s population to maintain health, and to prevent or treat diseases [10]. These bioactive constituents provide considerable opportunities for discovering antitumor drugs [2, 11]. Currently, most antitumor agents are generated from microorganisms, macroscopic organisms and plants, such as camptothecin and paclitaxel (Taxol®), two well-known anticancer drugs derived from plants [10, 12].
Brucea javanica (L.) Merr, which grows extensively throughout southern China and Southeast Asia, is an indeciduous plant shrub from the Simaroubaceae family [13]. The fruit of Brucea javanica ( Figure 1a, b ) is oval-shaped and solid, with a typical width and length of approximately 5 mm and 8 mm, respectively [14]. According to the official Chinese Pharmacopoeia describing traditional Chinese medicine (TCM) components, the fruit of Brucea javanica is generally used to combat many diseases, including diarrhea, malaria, intestinal inflammation and various types of cancer [15, 16]. In China, Bruceae Fructus oil is used for the clinical management of esophageal carcinoma, lung cancer and hepatocellular carcinoma, without toxic effects, thus improving immune regulatory function and patient quality of life [17–20].

The origin of brusatol.
a) The fruits of Brucea javanica (from The Plants in Shaoguan National Forest Park, ISBN: 978-7-5219-0150-4). b) The sundried fruits of Brucea javanica. c) The structure of brusatol.
Brusatol ( Figure 1c ), a primary natural component of Bruceae Fructus, is considered one of the bioactive bases necessary for the antineoplasm property of Bruceae Fructus [21]. This product has various biological effects, such as insecticidal [22], antimalarial [23], anti-inflammatory [24] and anticolitis activities [25, 26]. Numerous efforts have been aimed at determining the antitumor properties of this component and investigating its potential as an antitumor drug.
Herein, we summarize the antitumor characteristics of brusatol and the potential signaling pathways through which it acts, as demonstrated by in vivo and in vitro experiments. We additionally summarize brusatol’s toxicology, pharmacokinetics and drug delivery tools. On the basis of previous studies on brusatol, we discuss several challenges and provide suggestions to promote the study of brusatol and provide a reference for future work.
2. RETRIEVAL AND ANALYSIS METHODS
A comprehensive search was performed to identify all articles related to the use of brusatol indexed by Web of Science (1994–present) and PubMed (1994–present). The only keyword used in the search was “brusatol”. After carefully reading all the identified articles, we compiled the details of the in vitro and in vivo experiments therein.
From all half-maximal inhibitory concentration (IC50) values reported in the previous articles, we selected the data for 72 hours of brusatol treatment; eliminated the data from problematic cell lines, as demonstrated in other articles; grouped the data according to cancer type; eliminated all groups with fewer than three total samples; and performed statistical analyses in Prism 7.0 software (GraphPad Software, USA). The results are shown as mean ± SEM.
3. ANTICANCER PROPERTIES OF BRUSATOL
3.1 In vitro studies
In in vitro experiments, brusatol exhibited the potential to arrest the cell cycle, promote apoptosis, induce autophagy, attenuate epithelial-mesenchymal transition (EMT), inhibit migration, invasion and angiogenesis, and increase the chemosensitivity and radiosensitivity of various cancer cell lines ( Table 1 ). The IC50 values of brusatol in cancer cell lines are shown in Figure 2 , in which mean values in various types of cancer indicate the sensitivity to brusatol. As shown in Figure 2 , the sensitivity of cancers to brusatol is as follows: lymphoma > leukemia > multiple myeloma > lung cancer > glioma > nasopharyngeal carcinoma (NPC) > skin cancer > ovarian cancer > kidney cancer > liver cancer > pancreatic cancer > breast cancer > colon cancer.
The effects of brusatol against cancer cells in in vitro experiments.
| Cancer | Cell Lines | Effect | Mechanism of Action | Ref | ||
|---|---|---|---|---|---|---|
| Breast cancer | BT-474 | ↑Chemosensitivity (trastuzumab), apoptosis | ↓Growth | ↑ROS | ↓Nrf2, HO-1, p-HER2/HER2, p-AKT/AKT, p-ERK1/2/ERK1/2 | [27] |
| Breast cancer | SK-BR-3 | — | ↓Growth | — | ↓Nrf2, HO-1, p-HER2/HER2, p-AKT/AKT, p-ERK1/2/ERK1/2 | [27] |
| Breast cancer | MCF-7 | ↑Apoptosis | ↓Viability | ↑ROS | ↓ΔΨm | [28] |
| Breast cancer | MCF-7 | ↑Mammosphere anchorage-independent chemosensitivity (paclitaxel) | ↓Mammosphere anchorage-independent growth | ↑ROS | ↓Nrf2, NQO1, GCLM, HO-1, GSH | [29] |
| Breast cancer | MDA-MB-231 | ↑Mammosphere anchorage-independent chemosensitivity (paclitaxel) | ↓Mammosphere anchorage-independent growth | ↑ROS | ↓Nrf2, NQO1, GCLM, HO-1, GSH | [29] |
| Breast cancer | MDA-MB-231 | ↑Chemosensitivity (cisplatin) | — | — | ↓ARE, Nrf2 | [30] |
| Breast cancer | MDA-MB-231 | ↑Apoptosis, chemosensitivity (paclitaxel) | ↓Viability, migration, invasion, EMT | ↑E-cadherin, ROS, Bax | ↓Vimentin, Bcl-2 | [31] |
| Breast cancer (murine) | 4T1 | ↑Chemosensitivity (plumbagin), oxidative stress, | ↓Viability | — | — | [32] |
| Cervical cancer | HeLa | ↑Chemosensitivity (cisplatin) | — | — | ↓Nrf2 | [30] |
| Colon cancer | DLD-1 | ↑Cell death, chemosensitivity (thiabismocine) | — | — | ↓NRF2, HO-1 | [33] |
| Colon cancer | CT-26 | ↑Apoptosis | — | ↑pro-Caspase-3, pro-Caspase-9 | ↓Bcl-2 | [34] |
| Colon cancer | CT-26 | ↑Chemosensitivity (cisplatin) | — | ↑Cyt c, pro-Caspase-3, pro-Caspase-9 | — | [34] |
| Colon cancer | HCT116 | ↑Chemosensitivity (irinotecan) | ↓Viability, colony formation | — | ↓Nrf2 | [35] |
| Colon cancer | CCD-33Co | — | ↓Viability | — | — | [35] |
| Colon cancer | RKO, HCT116 | ↑Cell death | — | ↑PHD1, PHD2, PHD3, Fe2+ | ↓HIF-1α, Glut1, PGK1, PDK1, CA9, mtROS, c-Myc | [36] |
| Colon cancer | SW480, CoLo205, DLD-1, HT29, HCT15 | — | — | — | ↓HIF-1α | [36] |
| Colon cancer | HCT116 | — | ↓Proliferation, glycolytic enzymes, glucose consumption | — | ↓HIF-1α, VEGF, GLUT1, HK2, LDHA, ROS, mt-ROS | [37] |
| Colon cancer (murine) | CT26 | ↑Chemosensitivity (irinotecan) | ↓Viability, colony formation | — | ↓Nrf2 | [35] |
| Endometrial cancer | ECC1# | ↑Chemosensitivity (plumbagin), oxidative stress, apoptosis | ↓Viability | — | — | [32] |
| Endometrial cancer | Ishikawa | ↑Chemosensitivity (progestin) | — | — | ↓Nrf2, AKR1C1 | [38] |
| Endometrial cancer | Ishikawa, Spec-2 | — | — | — | ↓Nrf2 | [30] |
| Gastric cancer | SGC-7901# | ↑Apoptosis | ↓Migration, invasion, EMT | ↑IL-10, Bax, cleaved Caspase-3, ROS | ↓N-cadherin, MMP-9, Vimentin, IL-17, p-PI3K/PI3K, p-AKT/AKT, NF-kB, Bcl-2, Caspase-3 | [39] |
| Glioma | IDH1-mutated U251 | ↑Nrf2 ubiquitination, oxidative damage | ↓Nrf2-associated gene transcription | ↑ROS | ↓Nrf2, GCLC, GCLM, SLC7A11, NQO1, GSH/GSSG | [40] |
| Glioma | U251 | ↑DNA damage | ↓Proliferation | ↑ROS, 8-OH-dG, 8-oxoG, γH2A. X, cleaved PARP | ↓Nrf2, HMOX1, NQO1, GCLC | [41] |
| Glioma | GSC627, GSC711 | — | ↓Sphere formation | — | — | [41] |
| Glioma | A172, U251, U87 | ↑Apoptosis | ↓Viability, colony formation, migration, invasion | ↑Bax, cleaved Caspase-3, cleaved Caspase-9 | ↓Bcl-2, pro-Caspase-3, pro-Caspase-9, ECM1 | [42] |
| Hypopharyngeal carcinoma | UD-SCC-2 | ↑Apoptosis | ↓Metastasis, angiogenesis, proliferation | ↑Cleaved Caspase-3, cleaved PARP | ↓p-STAT3, p-JAK1(Tyr1022/1023), p-JAK2(Tyr1007/1008), p-Src(Tyr416), Survivin, Bcl-2, Bcl-xl, COX-2, VEGF, Cyclin E, Cyclin D1 | [43] |
| Hypopharyngeal carcinoma | FaDu | ↑Apoptosis | ↓Metastasis, angiogenesis, proliferation | — | ↓Survivin, Bcl-2, Bcl-xl, COX-2, VEGF, Cyclin E, Cyclin D1 | [43] |
| Kidney cancer | A498, ACHN, OSRC-2 | ↑Apoptosis | ↓Viability, colony formation, migration, invasion | ↑PTEN, Bax, E-cadherin | ↓p-PI3K/PI3K, p-AKT/AKT, Bcl-2, N-cadherin, Vimentin, MMP-9, MMP-2 | [44] |
| Leukemia | MOLM14 | ↑G0/G1 cell-cycle arrest | — | — | — | [45] |
| Leukemia | THP1 | ↑Apoptosis, chemosensitivity (cytarabine, daunorubicin, arsenic trioxide) | ↓Colony formation | — | ↓Nrf2, NQO1, GCLC, GCLM | [46] |
| Leukemia | U937 | ↑Chemosensitivity (cytarabine, daunorubicin, arsenic trioxide) | — | — | — | [46] |
| Leukemia | HL-60 | ↑Cell differentiation | — | ↑p100, p65, p105, p50, pIkBa | ↓IkBa | [47] |
| Leukemia | HL-60 | ↑G0/G1 cell-cycle arrest, monocyte-like characteristics | — | ↑CD11b, CD13 | ↓CD15, c-MYC | [48] |
| Leukemia | K562 | ↑G0/G1 cell-cycle arrest, monocyte-like characteristics, erythrocytic differentiation | — | — | ↓CD15, c-MYC | [48] |
| Leukemia | Kasumi-1 | ↑G0/G1 cell-cycle arrest, monocyte-like characteristics | — | — | ↓c-MYC | [48] |
| Leukemia | NB4 | ↑Sub-G1 apoptotic peak, monocyte-like characteristics | — | ↑CD13 | ↓CD15, c-MYC | [48] |
| Leukemia | U937 | ↑Metabolic arrest (S phase), monocyte-like characteristics | — | ↑CD11b, CD13 | ↓CD15, c-MYC | [48] |
| Leukemia | BV173 | ↑G0/G1 cell-cycle arrest, metabolic arrest (S phase), monocyte-like characteristics, erythrocytic differentiation | — | — | ↓c-MYC | [48] |
| Leukemia | SUPB13 | ↑G0/G1 cell-cycle arrest, erythrocytic differentiation | — | — | ↓c-MYC | [48] |
| Leukemia | RS4;11 | ↑Metabolic arrest (S phase), erythrocytic differentiation | — | — | ↓CD15, c-MYC | [48] |
| Leukemia | Reh | ↑G0/G1 cell-cycle arrest | — | — | ↓c-MYC | [48] |
| Liver cancer | HCCLM3 | — | ↓Invasion, EMT | ↑Occludin, E-cadherin | ↓Fibronectin, Vimentin, N-cadherin, Twist, Snail, p-Stat3(Tyr705)/Stat3 | [49] |
| Liver cancer | Hep3B, Huh7, LM3 | ↑Autophagy | — | ↑LC3-II/I, Beclin1 | ↓p62 | [50] |
| Liver cancer | Bel7404# | ↑Apoptosis, autophagy | ↓Colony formation, EMT, invasion, migration | ↑Bax, cleaved Caspase-3, cleaved PARP, LC3-II/I, Beclin1, E-cadherin | ↓Bcl-2, p62, PI3K, p-AKT/AKT, p-mTOR/mTOR, N-cadherin, Vimentin | [50] |
| Liver cancer | Bel-7402# | ↑Apoptosis | ↓Viability | ↑ROS | ↓ΔΨm | [28] |
| Liver cancer | HepG2 | — | ↓Viability | — | ↓Nrf2, p-Nrf2, GCLC, GCLM, GR, rGSH/GSSG | [51] |
| Liver cancer (murine) | Hepa-1c1c7 | ↑Chemical toxicity (2,4-dinitrochlorobenzene, iodoacetamide, N-acetyl-p-benzoquinone imine) | — | — | ↓Nrf2, ATP | [52] |
| Lung cancer | PC9 | ↑Apoptosis, G0/G1 cell-cycle arrest | ↓Clonogenic growth, migration | ↑ROS, Bax, Bak, cleaved/pro-Caspase-3, cleaved/pro-Caspase-8, cleaved/total PARP | ↓Nrf2, HO-1, Bcl-2, Bcl-xl, XIAP, | [21] |
| Lung cancer | PC9 | ↑Chemosensitivity (paclitaxel, erlotinib, gefitinib, cisplatin) | — | — | — | [21] |
| Lung cancer | H460 | — | ↓EMT | ↑Vinculin | ↓Nrf2, RhoA, Rock1, β-catenin, Snail, Slug, E-cadherin, RhoA-GTP, F-actin | [53] |
| Lung cancer | A549 | — | ↓Migration, EMT | ↑β-catenin, Slug, Vinculin | ↓Nrf2, RhoA, Rock1, Snail, E-cadherin, RhoA-GTP | [53] |
| Lung cancer | HCC827GRKU | ↑G0/G1 cell-cycle arrest | ↓Viability, migration, invasion | ↑HMOX1 | ↓NRF2, AKR1C1, AKR1C2, AKR1C3, AKR1B1, AKR1B10, GCLC, GCLM, HO1 | [54] |
| Lung cancer | HCC827GRKU | ↑Chemosensitivity (gefitinib, afatinib, osimertinib) | — | — | — | [54] |
| Lung cancer | A549 | — | ↓Cap-dependent and cap-independent translation | — | ↓Nrf2, p53, GFP, p21 | [55] |
| Lung cancer | A549 | — | ↓Viability | — | ↓Nrf2, GCLC | [56] |
| Lung cancer | A549 | ↑Radiosensitivity (Ionizing radiation), DNA damage | ↓Viability | ↑ROS | ↓Nrf2 | [57] |
| Lung cancer | H1299 | — | ↓Viability | — | — | [57] |
| Lung cancer | A549 | ↑Nrf2 ubiquitination, chemosensitivity (cisplatin), intracellular concentration of cisplatin | ↓Colony formation | — | ↓ARE, Nrf2, γGCS, MRP1, MRP2, NQO1, GSTM2, GCLC, GSH | [30] |
| Lung cancer | H1299, A549 | ↑G0/G1 cell-cycle arrest | ↓Viability, colony formation, migration, invasion | ↑E-cadherin, p27 | ↓Skp1 | [58] |
| Lymphoma | Raji | ↑G0/G1 cell-cycle arrest | — | ↑BAX | ↓AKT1, ATF3, PIK3C2B, JUP, NME2, PI3Kγ, P53, GSK3, CCND1, mTOR | [45] |
| Lymphoma | SU-DHL-4 | ↑G0/G1 cell-cycle arrest | — | — | ↓mTOR, AKT1, GSK3 | [45] |
| Lymphoma | Daudi## | — | — | — | ↓c-MYC | [48] |
| Multiple myeloma | MM.1S, L363, U266 | ↑Oxidative stress | ↓Viability, growth | — | — | [59] |
| Nasopharyngeal carcinoma | CNE-1# | ↑Apoptosis, cell-cycle arrest | ↓Proliferation | ↑Bad, Bax, cleaved PARP, cleaved Caspase-7, cleaved Caspase-3, p-Cdc2 | ↓Bcl-xl, Bcl-2, PARP, Caspase-9, Caspase-7, Caspase-3, Cdc25c, Cyclin B1, Cdc2 p34, Cyclin D1, p-AKT/t AKT, p-mTOR/t mTOR, p-4EBP1/t 4EBP1, p-S6K | [60] |
| Oral cancer | JMAR#, YD-10B | ↑Apoptosis | ↓Metastasis, angiogenesis, proliferation | ↑Cleaved Caspase-3, cleaved PARP | ↓p-STAT3, p-JAK1(Tyr1022/1023), p-JAK2(Tyr1007/1008), p-Src(Tyr416), Survivin, Bcl-2, Bcl-xl, COX-2, VEGF, Cyclin E, Cyclin D1 | [43] |
| Oropharyngeal carcinoma | LN686 | ↑Apoptosis | ↓Metastasis, angiogenesis, proliferation | — | ↓Survivin, Bcl-2, Bcl-xl, COX-2, VEGF, Cyclin E, Cyclin D1 | [43] |
| Ovarian cancer | SK-OV-3 | ↑Chemosensitivity (trastuzumab), apoptosis | ↓Growth | ↑ROS | ↓Nrf2, HO-1, p-HER2/HER2, p-AKT/AKT, p-ERK1/2/ERK1/2 | [27] |
| Ovarian cancer | SKOV3 | ↑Chemosensitivity (plumbagin) | ↓Viability | — | — | [32] |
| Ovarian cancer | OVCAR3 | ↑Chemosensitivity (plumbagin), oxidative stress, apoptosis | ↓Viability | — | — | [32] |
| Ovarian cancer | A2780CP, CoC1/DDP | ↑Chemosensitivity (cisplatin) | — | ↑SLC40A1 | ↓Nrf2 | [61] |
| Pancreatic cancer | PATU-8988 | ↑Apoptosis | ↓Growth | ↑ROS, Bax, cleaved Caspase-3 | ↓MPR1, MPR2, MPR3, MPR4, MPR5, HO-1, NQO1, Nrf2, Bcl-2 | [62] |
| Pancreatic cancer | PANC-1## | ↑Apoptosis | ↓Growth | ↑Bax, cleaved Caspase-3 | ↓MPR4, MPR5, HO-1, NQO1, Nrf2, Bcl-2 | [62] |
| Pancreatic cancer | BxPC-3## | ↑Apoptosis | ↓Growth | ↑ROS, Bax, cleaved Caspase-3 | ↓MPR1, MPR2, MPR4, MPR5, HO-1, NQO1, Nrf2, Bcl-2 | [62] |
| Pancreatic cancer | PANC-1##, Capan-2## | ↑Chemosensitivity (gemcitabine, 5-fluorouracil), apoptosis, G2/M cell-cycle arrest | ↓Proliferation | ↑E-cadherin | ↓NF-κB p65, Bcl-xL, PCNA, Vimentin, Twist | [63] |
| Pancreatic cancer | Capan-1, SW1990 | — | ↓Proliferation | — | — | [63] |
| Pancreatic cancer | PATU-8988 | ↑Apoptosis | ↓Viability, proliferation | ↑Bax, cleaved Caspase-3, p-JNK, p-c-Jun, p-p38 | ↓Bcl-2, Caspase-3, p-p65, p65, p-Stat3 | [64] |
| Pancreatic cancer | PANC-1## | — | — | ↑Bax, cleaved Caspase-3, p-JNK, p-c-Jun, p-p38 | ↓Bcl-2, Caspase-3, p-p65, p65, p-Stat3 | [64] |
| Pheochromocytoma | hpheo1 | — | — | — | ↓NRF2, xCT, GCLC, GCLM, SLC7A11 | [65] |
| Pheochromocytoma (murine) | MPC | ↑DNA damage, apoptosis | ↓ROS homeostasis, Colony formation | ↑ROS, Caspase 3/7, cleaved PARP, γH2A. X, | ↓NRF2, xCT, GCLC, GCLM, SLC7A11 | [65] |
| Pituitary adenoma(murine) | GH3, MMQ | ↑Apoptosis, chemosensitivity (cabergoline) | ↓Viability, hormone secretion, colony formation | ↑Cleaved Caspase-3, cleaved Caspase-8, ROS | ↓Bcl-2, p-S6K1/t S6K1, p-4EBP1/t 4EBP1, Nrf2, HO-1 | [66] |
| Skin cancer | A375 | ↑Phototherapy, G0/G1 cell-cycle arrest, apoptosis | ↓Growth, proliferation, colony formation | ↑ROS, COX2, CDK2, Bax, cleaved Caspase-3 | ↓Nrf2, GSTP1, CCND1, CCNE2, CDK4, CDK6, Bcl-2, Bcl-xl, HO-1, p-AKT(Ser473) | [67] |
# Problematic cell line; ## Caution.

3.1.1 Cell-cycle arrest
The cell cycle, an essential process of cell division, consists primarily of four phases: pre-DNA synthesis (G1 phase), DNA synthesis (S phase), late DNA synthesis (G2 phase) and mitosis (M phase). Cyclins, cyclin-dependent kinases (CDKs) and cyclin-dependent-kinase inhibitors have important roles in cell-cycle regulation. CDKs, whose activity is dependent on their corresponding cyclins, promote cell-cycle progression by phosphorylating specific substrates. However, cyclin-dependent-kinase inhibitors bind CDKs, thus inhibiting their kinase activity, and negatively regulate the cell cycle [68–70]. Cancer initiation or progression results from aberrant expression of various cell-cycle proteins. Therefore, regulating the cell cycle is becoming a promising strategy in cancer therapy [71, 72].
Brusatol induces cell-cycle arrest at G0/G1, S or G2/M phase, depending on the cell-line characteristics rather than the cancer type. Among leukemia cell lines, brusatol induces G0/G1 cell-cycle arrest in Kasumi-1, K562, HL-60, Reh, SUPB13 and BV173 cells, but causes metabolic arrest (S phase) in U937 and RS4 cells [11]. Brusatol also simultaneously induces arrest at two phases (G0/G1 and S) in the leukemia cell line BV173 [48]. Similar results have also been found in NPC (CNE-1), with a decrease in cyclin B1, cyclin D1, Cdc2, and Cdc25c, and an increase in p-Cdc2; the cells also are arrested in only one phase [60]. In other cancer types, brusatol exerts an anti-proliferative effect by interfering with the cell cycle, including G0/G1 phase in the PC9 and HCC827GRKU lung cancer cell lines [21, 54], and the Raji and SU-DHL-4 lymphoma cell lines [45], and G2/M phase in the pancreatic cancer lines Capan-2 and PANC-1 [63]. In A375 melanoma cells, Wang et al. have found that cyclin D1, cyclin E2, CDK4 and CDK6 decrease after brusatol treatment, thus causing G0/G1 cell-cycle arrest [67]. These findings suggest that brusatol inhibits the cell cycle in multiple tumor cell types and thereby inhibits the proliferation of tumor cells.
According to previous studies, the inhibitory effects of brusatol on the cell cycle should occur in most stages, but the sensitivity of different stages of the cell cycle to the inhibitory effect of brusatol differs among cancer cells. The inhibitory effect of brusatol is dominant at specific stages in various cancer cells, whereas all highly expressed proteins associated with the cell cycle may be inhibited.
3.1.2 Apoptosis
In multicellular organisms, apoptosis, or programmed cell death, has a major role in maintaining cellular homeostasis [73]. Apoptotic pathways consist of two main pathways: the mitochondrial pathway (intrinsic pathway) and the death-receptor-mediated pathway (extrinsic pathway) [74]. The mitochondrial pathway is associated with altered mitochondrial membrane permeability (MMP), thus resulting in an imbalance between Bak and Bax, and the release of mitochondrial cytochrome C (Cyt C) together with other related proteins. The released Cyt C binds apoptosis protease activating factor 1 (Apaf1) and subsequently induces the formation of the apoptosome complex, which activates caspase-9 and later facilitates the downstream activation of caspase-3 and caspase-7, thus executing cell death. In the extrinsic pathway, the binding of ligands, including Fas ligand (Fas-L), tumor necrosis factor (TNF) and TNF-associated apoptosis-inducing ligand, to their corresponding receptors activates caspase-8 and induces apoptosis [73–77]. Notably, cancer cells avoid apoptosis on their path to becoming cancerous. Hence, a major strategy in cancer therapy is promoting cancer cell apoptosis [78].
Brusatol initiates caspase-dependent apoptotic pathways in various cancer types. Zhang et al. have found that MCF-7 human breast cancer cells lose their MMP after treatment with 5 μM brusatol, thus causing apoptosis, as also confirmed in Bel-7402 human liver cancer cells [28]. Other studies have concluded that brusatol causes a substantial imbalance between Bcl-2 and Bax [21, 39, 50, 60, 62, 64, 67]. The increasing Bax/Bcl-2 ratio results in the disengagement of Cyt C together with other related proteins, thus promoting the caspase cascade and apoptosis. Beyond this mitochondrially mediated pathway (intrinsic pathway), brusatol targets two TNF-associated apoptosis-inducing ligand receptors—tumor necrosis factor receptor and death receptor—thus causing caspase-8 and caspase-3 activation, target-protein cleavage and ultimately apoptosis [21].
3.1.3 Autophagy
Physiologically, autophagy is a common process in which cells under conditions of starvation or energy deficiency synthesize new ATP and macromolecules via a series of reactions [79]. Autophagy is regulated by the mTORC complexes mTORC1 and mTORC2, particularly mTORC1, whose activation causes autophagy-associated protein (ATG) phosphorylation and subsequent autophagy inhibition [80, 81]. The inhibition of mTORC1 results in activation of the Unc-51-like autophagy-activating kinase complex, thus causing the phagophore to colocalize with activated class III phosphatidyl-inositol 3-kinase (PI3K) [82, 83]. Beclin-1 has an important role in suppressing cancer, through recruiting autophagosome elongation- and maturation-associated proteins [84]. ATGs also have a major role in regulating autophagosome elongation. Microtubule-associated protein 1 light chain 3 II (LC3 II) contributes to the binding of autophagosomes to degraded substrates; subsequently, autophagosomes selectively remove damaged organelles via lysosomes. ATG3 and ATG7 disorders interfere with the conversion of LC3 I to LC3 II and the processes described above [85–87]. Autophagy combats various cancers by contributing to cancer cell death, thus making it a potent target for cancer treatment [88].
The potential of brusatol to activate protective autophagy has been demonstrated in hepatocellular carcinoma (HCC) cell lines via the conversion of LC3 I to LC3 II, in a process involving autolysosome formation and apoptosis. In Bel7404 cells, the PI3K/AKT/mammalian target of rapamycin (mTOR) signaling pathway is inactivated by brusatol, thus causing cytoprotective autophagy. This brusatol-treated HCC cell line expresses less p62 but more Beclin-1 than control cells [50]. These studies have offered numerous lines of evidence of the effect of brusatol in inducing cancer cell autophagy.
3.1.4 Epithelial-mesenchymal transition
EMT is a process during which epithelial-like cells transform into migratory mesenchymal-like cells, which participate in tissue fibrosis, tumor invasiveness and metastasis. From the perspective of therapeutic failures in cancer, the plasticity of this cellular characteristic offers a novel strategy [89, 90]. During EMT, mesenchymal proteins (N-cadherin and vimentin) increase, and epithelial proteins (cytokeratin and E-cadherin) are lost [91]. In addition, several crucial transcription factors (ZEB1/2, Snail1, Snail2 and Twist) have major roles in EMT [89, 92].
Brusatol has been shown to suppress the EMT process in various types of tumors, including gastric cancer, liver cancer, and lung cancer ( Table 1 ). Brusatol suppresses the expression of mesenchymal proteins such as N-cadherin and vimentin as well as the matrix metalloproteinase MMP-9, thus inhibiting the invasion of gastric cancer cells [39]. In addition, brusatol induces the expression of the epithelial marker E-cadherin and the tight-junction protein occludin, and inhibits fibronectin and two key transcription factors, Twist and Snail (Snail 1), thus suppressing EMT in the HCCLM3 HCC cell line [49]. Brusatol also decreases the expression of Slug and its target gene β-catenin in lung cancer. However, the expression of Slug and β-catenin increases in A549 lung cancer cells treated with brusatol [53].
3.1.5 Migration, invasion and angiogenesis
Metastasis, which involves invasion and migration, is the leading cause of death in patients with cancer. Nearby tissues are the sites of cancer cell migration and invasion, and cancer cells reach distant organs by entering the lymphatic or circulatory system [93–95]. Invadopodia of cancer cells, which are formed after stimulation by epidermal growth factor, are essential for remodeling membrane proteins and extracellular matrix, thus promoting metastasis [93]. Hence, during malignancy treatment, preventing the metastasis of cancer cells is a major goals particularly for patients in early disease stages [95].
Numerous studies have shown that brusatol inhibits metastasis of various types of cancer, such as gastric cancer [39], hypopharyngeal carcinoma [43], liver cancer [50, 49], lung cancer [21, 53, 54], oral cancer [43] and oropharyngeal carcinoma [43]; this inhibition is often accompanied by a loss of epithelial markers and an increase in mesenchymal markers. Furthermore, brusatol inhibits the expression of MMP-9 in gastric cancer cells, which has a major role in the cancer metastatic process [39].
Angiogenesis, the formation of new blood vessels, supplies nutrients and oxygen for cells and tissues, and has a major role in the development and progression of malignancies [96]. With the release of proteins promoting cellular growth and motility, endothelial cells in developing cancer are stimulated and subsequently build a network of blood vessels for the alleviation of hypoxic conditions; vascular endothelial growth factor (VEGF) and fibroblast growth factor-2 (FGF2) have core roles in this process [97, 98].
In oral cancer cell lines (JMAR and YD-10B), brusatol downregulates the expression of VEGF via the STAT3 pathway, thus inhibiting angiogenesis, as also confirmed in hypopharyngeal carcinoma cells (UD-SCC-2 and FaDu) and oropharyngeal carcinoma cells (LN686) [43].
3.1.6 Chemosensitivity and radiosensitivity
In rapidly dividing cells, radiation causes the cleavage of water molecules and the generation of reactive oxygen species (ROS), which directly damage DNA; thus, radiotherapy is often used in cancer treatment [99]. Several factors affect the radiosensitivity of cancer cells, such as therapy-altered pathways, the expression of proto-oncogenes or anti-oncogenes, and the repair of DNA damage [100–102]. Drugs targeting these pathways are potential radiosensitizers to tumor therapy, and numerous studies have indicated that during cancer radiotherapy, brusatol markedly improves their effects.
Some researchers have observed that brusatol promotes ROS production and enhances DNA damage through the inhibition of Nrf2 expression in A549 lung cancer cells in vitro when combined with radiotherapy [57]. Moreover, brusatol inhibits the expression of Nrf2, p-ATR and p-CHK1, thus increasing radiosensitivity [103].
Because of adverse events and the evolution of drug resistance, chemotherapy options are limited for patients with cancer. Thus, decreasing the adverse effects or increasing the sensitivity to chemotherapy is an important strategy for cancer therapy. In China, TCM herbal therapy is considered a promising complementary therapy, because of its doctrine of individualized therapy and a holistic approach to treating various syndromes. In addition, it has numerous favorable effects for patients with cancer, such as prolonging overall survival, improving quality of life and decreasing adverse events. Chemotherapy combined with TCM therapy for patients with cancer has gained increasing attention because TCM therapy places a greater focus on overall functional adjustment and bodily recovery than chemotherapy [104]. Numerous studies have demonstrated that chemotherapy combined with TCM therapy prolongs overall survival in patients with cancer by mitigating the adverse effects of chemotherapy and enhancing immunity; thus, use of the bioactive components extracted from TCMs in combination therapies to decrease the toxicity and overcome the limitations of chemotherapy is attracting increasing attention [105–108].
Several studies have demonstrated that chemotherapy combined with brusatol enhances clinical effects and decreases adverse events. Many studies have shown that brusatol increases the effects of many chemical drugs, including trastuzumab, paclitaxel, cisplatin (CDDP), plumbagin, thiabismocine, irinotecan, progestin, cytarabine, daunorubicin, arsenic trioxide, 2,4-dinitrochlorobenzene, iodoacetamide, N-acetyl-p-benzoquinone imine, erlotinib, gefitinib, afatinib, osimertinib, gemcitabine and 5-fluorouracil [21, 27, 29, 30–35, 38, 46, 54, 63].
Moreover, in the human melanoma cell line A375, brusatol significantly increases the effects of phototherapy by increasing ROS-induced cell-cycle arrest and cellular apoptosis, and inhibiting melanoma growth through the AKT/Nrf2 pathway [67].
3.2 In vivo studies
In in vivo animal models, brusatol inhibits tumor growth, invasion and metastasis, and promotes apoptosis, radiosensitivity and chemosensitivity ( Table 2 ).
The anticancer effects of brusatol in in vivo tumor-bearing animal models.
| Cancer | Cell Lines | Observation | Mechanism of Action | Ref | ||
|---|---|---|---|---|---|---|
| Breast cancer | BT-474 | ↑Chemosensitivity (trastuzumab) | ↓Tumor growth | — | ↓Nrf2, HO-1, p-HER2/HER2, p-AKT/AKT, p-ERK1/2/ERK1/2 | [27] |
| Colon cancer | RKO, HCT116 | — | ↓Tumor growth | ↑TUNEL | ↓HIF-1α, c-Myc | [36] |
| Colon cancer (murine) | CT26 lucA6c | ↑Chemosensitivity (irinotecan) | ↓Tumor growth | — | ↓Nrf2 | [35] |
| Glioma | TS603 | — | ↓Tumor growth | ↑γH2A. X, TUNEL | ↓Ki67, Nrf2, SLC7A11, GCLC, GCLM | [40] |
| Glioma | TS603 | — | ↓Tumor growth | ↑8-oxoG, γH2A. X, TUNEL | ↓Nrf2, NQo1, GCLC, Ki67, PCNA | [41] |
| Glioma | U87 | — | ↓Tumor growth, Invasion | ↑TIMP1, TIMP2 | ↓ECM1, MMP1, MMP2, MMP9 | [42] |
| Leukemia | MOLM14 | — | ↓Tumor growth | — | — | [45] |
| Liver cancer | H22 | ↑Apoptosis, overall survival | — | ↑miRNA-29b gene, p53, Bax, Bad, Cyto c, cleaved Caspase-9, cleaved Caspase-3, PARP, cleaved PARP | ↓Bcl-2 | [109] |
| Liver cancer | HCCLM3-Luc | — | ↓Tumorigenesis, metastasis | ↑E-cadherin, Occludin | ↓Ki-67, Vimentin, Twist, Fibronectin, N-cadherin, Snail | [49] |
| Liver cancer | Bel7404# | — | ↓Tumor growth | — | ↓Ki67, MMP2, MMP9 | [50] |
| Lung cancer | A549 | ↑Radiosensitivity (ionizing radiation) | ↓Tumor growth | — | ↓p-ATR(Ser428), NRF2, p-CHK1(Ser317) | [103] |
| Lung cancer | HCC827GKRU | — | ↓Tumor growth, Invasion | — | — | [54] |
| Lung cancer | — | ↑Cancer initiation, proliferation, oxidative DNA damage | — | ↑NRF2, GCLM, NQO1, AKR1C1, AKR1B10, p-ERK, HMOX1, Ki67 | ↓γ-H2AX, 8-oxo-dG | [110] |
| Lung cancer | — | ↑Apoptosis | ↓Cancer progression, proliferation, oxidative DNA damage | ↑γ-H2AX, 8-oxo-dG, TUNEL | ↓NRF2, GCLM, NQO1, AKR1C1, AKR1B10, p-ERK, HMOX1, Ki67, | [110] |
| Lung cancer | — | ↑Chemosensitivity (cisplatin), DNA damage, apoptosis | ↓Tumor growth, proliferation | ↑γ-H2AX, 8-oxo-dG, TUNEL | ↓Nrf2, Akr1b10, Akr1c1, Nqo1, Gclm, Hmox1, Ki67 | [111] |
| Lung cancer | A549 | ↑Chemosensitivity (cisplatin) | ↓Nrf2 in xenografts | ↑TUNEL | ↓Ki67 | [30] |
| Lung cancer | A549-V or A549-K | ↑Chemosensitivity (cisplatin) in an Nrf2-dependent manner | — | ↑TUNEL in an Nrf2-dependent manner | ↓Ki67 in an Nrf2-independent manner | [30] |
| Lung cancer | A549 | — | ↓Tumor growth, Metastasis | ↑E-cadherin, p27 | ↓Ki-67, Skp1, Skp2 | [58] |
| Melanoma | A375 | — | ↓Tumor growth | — | — | [67] |
| Nasopharyngeal carcinoma | CNE-1# | — | ↓Tumor growth | ↑TUNEL | ↓Ki67 | [60] |
| Ovarian cancer | SK-OV-3 | ↑Chemosensitivity (trastuzumab) | ↓Tumor growth | — | ↓Nrf2, HO-1, p-HER2/HER2, p-AKT/AKT, p-ERK1/2/ERK1/2 | [27] |
| Pancreatic cancer | PANC-1## | ↑Chemosensitivity (gemcitabine) | ↓Tumor growth | ↑Cleaved Caspase-3 | ↓Nrf2, NQO1, Ki67 | [62] |
| Pancreatic cancer | Capan-2##, PANC-1## | ↑Chemosensitivity (gemcitabine, 5-fluorouracil) | ↓Tumor growth | ↑E-cadherin | ↓Twist | [63] |
| Pheochromocytoma (murine) | MPC SDHBKD Luc | ↑Overall survival | ↓Liver metastasis | ↑γH2A. X, TUNEL | ↓NRF2, xCT, GCLM, Ki67 | [65] |
| Pituitary adenoma (murine) | GH3, MMQ | ↑Chemosensitivity (cabergoline) | ↓Tumor growth | — | ↓p-S6K1/t S6K1, p-4EBP1/t 4EBP1 | [66] |
# Problematic cell line; ## Caution.
In tumor suppression, brusatol has shown substantial effects against various cancers in vivo, at doses of 0–4 mg/kg through different injection routes, including injection into the abdominal cavity or around tumors.
Many studies have shown that brusatol inhibits tumor growth by downregulating the expression of the Ki67 protein and upregulating the number of TUNEL-positive cells in colon cancer [36], glioma [40, 41], liver cancer [50], lung cancer [30], NPC [60] and pancreatic cancer [62]. Interestingly, pretreatment with brusatol contributes to cancer initiation, whereas posttreatment with brusatol prevents cancer progression in vinyl carbamate-induced carcinogenesis A/J mice.
For years, chemo- and radio-resistance have weakened the responses of tumors to therapy. Brusatol enhances the radiotherapy effect of ionizing radiation [103] and the chemotherapy effects of trastuzumab [27], irinotecan [35], CDDP [30, 111], gemcitabine [62, 63] and 5-fluorouracil [63].
Danio rerio (zebrafish), owing to their large broods, rapid maturation time and small size, can complement studies in traditional cell models and mice, and have recently become a major cancer model. The transparent body wall allows for visualization of tumor progression, and makes experiments less time- and labor-consuming [112, 113]. Zebrafish embryos and juvenile zebrafish are widely used as powerful tools to study cancer invasion, metastasis and tumor-induced angiogenesis. According to Park et al., brusatol decreases the number of lung cancer cells in drug-treated embryos and the migration of lung cancer in the body. Brusatol, compared with a control treatment, has been found to decrease the area of cancer cell penetration in the vessels of the zebrafish larvae by 55%, thus inhibiting cancer cell migration, invasiveness and metastasis [54].
4. EFFECTS OF BRUSATOL IN VARIOUS CANCERS
Numerous preclinical studies have provided evidence of the therapeutic potential of brusatol and have demonstrated its roles in regulating various cancer hallmarks, such as proliferation, apoptosis, survival, invasion, metastasis and angiogenesis ( Tables 1 and 2 ). The effects of brusatol in various cancers and its potential regulatory mechanisms toward various proteins associated with various malignancies are briefly summarized below.
4.1 Breast cancer
Breast cancer in women has surpassed lung cancer and become the dominant cause of cancer worldwide: in 2020, approximately 2,300,000 new cases occurred, accounting for 11.7% of all cancer-related cases; moreover, breast cancer became the fifth-leading cause of global cancer-associated death, with 685,000 deaths [114]. Zhang et al. have demonstrated that brusatol has excellent potential to promote apoptosis and inhibit cell viability via an oxidation pathway, and this process is significantly enhanced by redox-sensitive micelles [28]. In vivo and in vitro studies of breast cancer have indicated that brusatol displays antitumor effects by inhibiting the HER2/AKT/ERK1/2 and Nrf2/HO-1 pathways. Simultaneously, brusatol markedly enhances the effect of trastuzumab by increasing ROS accumulation and apoptosis [27]. Similar studies have also shown that brusatol enhances the effects of CDDP and paclitaxel through the inhibition of the Nrf2-mediated defense mechanism, thus suggesting that brusatol may be developed into an adjuvant chemotherapy drug [30, 31]. In the murine ovarian cancer cell line 4T1, brusatol enhances the cytotoxic activity of plumbagin through inhibition of the Nrf-2-mediated antioxidative response and mitochondrial electron transport [32]. Wu et al. have demonstrated that brusatol can be used as a novel chemotherapy-adjuvant drug against refractory tumor-initiating cancer stem cells [29].
4.2 Cervical cancer
In women, cervical cancer ranked fourth in terms of both incidence and cancer-associated mortality in 2020, with approximately 604,000 new cases and 342,000 deaths worldwide [114]. Ren et al. have demonstrated that brusatol enhances the effect of CDDP through inhibition of Nrf2 expression in HeLa cells [30].
4.3 Colon cancer
Colorectal cancer was the third most frequently diagnosed cancer but the second-leading cause of cancer-associated death in 2020, with more than 1,900,000 new cases (including anal cancer) and 935,000 deaths, thus accounting for approximately one-tenth of cancer-associated cases and deaths [114]. Lu et al. have reported that brusatol decreases glucose consumption and downregulates the expression of HIF-1α under hypoxia or CoCl2-induced hypoxia in a concentration-dependent manner in the colon cancer cell line HCT116 without substantial cytotoxicity, thus indicating that brusatol regulates HIF-1α and suggesting its therapeutic potential in colon cancers [37]. In another study, a combination of brusatol and CDDP has shown synergistic inhibitory effects on cell proliferation and stimulatory effects on cell apoptosis in CT-26 colon cancer cells [34]. Through both in vivo and in vitro experiments, numerous studies have indicated that brusatol inhibits the c-Myc/ROS signaling pathway, increases HIF-1α through promoting prolyl hydroxylase (PHD) activity and ultimately induces colon cancer cell death under hypoxia [36]. The anticancer effect of brusatol has also been demonstrated in a syngeneic orthotopic mouse model of colorectal cancer [35].
4.4 Endometrial cancer
In women, uterine corpus cancer ranked sixth in terms of incidence in 2020, with 417,000 new cases and 97,000 deaths worldwide; the majority of these cancers were adenocarcinomas arising from the endometrium [114]. In the endometrial cancer cell lines Ishikawa and Spec-2, brusatol decreases Nrf2 protein levels in a concentration-dependent manner [30]. Kapur et al. have reported that brusatol enhances the cytotoxic activity of plumbagin by alleviating oxidative stress [32]. In addition, brusatol decreases progestin resistance through suppression of the Nrf2/AKR1C1 pathway [38].
4.5 Gastric cancer
Gastric cancer ranked fifth in terms of incidence and fourth in terms of mortality, and remained a major cancer in 2020, with more than 1,000,000 new cases and approximately 769,000 deaths worldwide (equating to 1 in 13 deaths) [114]. Chen et al. have reported that brusatol reverses lipopolysaccharide-induced EMT and induces apoptosis through the PI3K/Akt/nuclear factor kappa-B (NF-κB) pathway in human SGC-7901 gastric cancer cells [39].
4.6 Glioma
Brain cancer accounts for an estimated 3% of all cancer- associated cases worldwide [115]. In IDH1-mutated glioma, brusatol exhibits a potent tumor suppressive effect both in vitro and in vivo [40]. In a similar in vivo study, brusatol has been found to selectively inhibit IDH1-mutated cancer progression [41]. RNA-seq analysis has indicated that brusatol downregulates the expression of extracellular matrix protein 1 (ECM1), thus inhibiting the proliferation and invasion of the glioblastoma cell lines A172, U251 and U87 [42].
4.7 Head and neck squamous cell carcinoma
Head and neck squamous cell carcinoma (HNSCC) has very poor prognosis, and its incidence is increasing rapidly each year [116–118]. Lee et al. have demonstrated that brusatol is a potential blocker of the STAT3 pathway in various HNSCC cell lines, by decreasing STAT3 activation through blocking upstream kinases including Src, JAK1 and JAK2, and decreasing STAT3’s nuclear levels and ability to bind DNA [43].
4.8 Kidney cancer
Brusatol significantly inhibits proliferation, migration and invasion, and increases apoptosis of the renal cell carcinoma lines A498, ACHN and OSRC-2 [44].
4.9 Leukemia
Pei et al. have reported that brusatol arrests cells at G0/G1 phase in vitro and suppresses cancer growth in vivo [45]. Mata-Greenwood et al. have shown that brusatol induces leukemic cell differentiation and G1 cell-cycle arrest, which are associated with the downregulation of c-myc [48]. In a similar study, Cuendeta et al. have demonstrated that brusatol-induced leukemic cell differentiation involves NF-κB activation in the HL-60 cell line [47]. In addition, Karathedath et al. have reported that brusatol in combination with chemotherapeutic agents such as cytarabine, daunorubicin and arsenic trioxide modulates drug resistance in acute myeloid leukemia [46].
4.10 Liver cancer
Primary liver cancer ranked sixth in terms of incidence and third in terms of cancer-associated mortality in 2020, with an estimated 906,000 new cases and 830,000 deaths worldwide [114]. Zhang et al. have shown that brusatol decreases cancer cell viability via the Nrf2/ARE signaling pathway [51]. In another study, Zhang et al. have shown that brusatol exerts its antitumor effect via an oxidation pathway [28]. Ye et al. have reported that brusatol exerts its anticancer effects via the PI3K/Akt/mTOR pathway and subsequently inhibits cell viability and promotes autophagy-induced apoptosis [50]. In addition, Lee et al. have reported that brusatol suppresses STAT3-driven metastasis via downregulation of EMT both in vitro and in vivo [49]. Furthermore, brusatol prolongs the survival of H22 ascites tumor-bearing mice by inducing H22 cell apoptosis [109]. In mouse Hepa-1c1c7 liver cancer cells, brusatol provokes a rapid and transient depression of Nrf2 expression, and sensitizes cells to chemical toxicity [52].
4.11 Lung cancer
Lung cancer ranked second in terms of incidence and first in terms of cancer-associated mortality in 2020, with approximately 2,200,000 new cases and 1,800,000 deaths, accounting for an estimated one in ten (11.4%) cancers diagnosed and one in five (18.0%) deaths [114]. Xie et al. have demonstrated that the process through which brusatol inhibits proliferation in the PC9 cell line might be intimately associated with the inhibition of the Nrf2-mediated antioxidant response and modulation of the ROS-mediated mitochondrial-dependent pathway [21]. Park et al. have demonstrated that brusatol inhibits cancer cell proliferation and reverses gefitinib resistance and EGFR-TKI cross-resistance through inhibition of Nrf2 activity [54]. In another study, brusatol has been found to enhance the effect of chemotherapy via the inhibition of the Nrf2-mediated defense mechanism both in vitro and in vivo [30]. Sun et al. have reported that brusatol enhances the radiosensitivity of the A549 cell line through promoting ROS production and DNA damage [57]. Ko et al. have reported that brusatol decreases cell motility through RhoA/ROCK1 signaling [53]. Recently, using streptavidin-based immunoprecipitation and mass spectrometry analyses, Xing et al. have revealed that brusatol specifically targets Skp1, thus inhibiting lung cancer growth and metastasis [58].
4.12 Lymphoma and multiple myeloma
Brusatol arrests lymphoma cells at G0/G1 by specifically targeting the PI3K/AKT pathway [45]. In addition, brusatol suppresses cell viability and growth by increasing oxidative stress in multiple myeloma cells [59].
4.13 Nasopharyngeal carcinoma
NPC, a common malignancy in Southeast Asia and southern China, remains a major cause of cancer-associated death globally, with an incidence of approximately 50,000 deaths every year [119, 120]. On the basis of in vivo and in vitro experiments, brusatol induces apoptosis and inhibits growth in NPC cells via the Akt/mTOR/S6K/4EBP1 pathway [60].
4.14 Ovarian cancer
Brusatol enhances the cytotoxic activity of plumbagin in the ovarian cancer cell lines OVCAR3 and SKOV3 [32]. In a similar study, brusatol has been found to enhance the anticancer ability of trastuzumab both in vitro and in vivo [27]. In addition, brusatol decreases CDDP resistance via suppression of the iron export-associated gene SLC40A1 in CoC1/DDP and A2780CP ovarian cancer cells [61].
4.15 Pancreatic cancer
Pancreatic cancer, because of its poor prognosis in both men and women, ranked seventh in terms of cancer-associated mortality in 2020, with approximately 496,000 cases and 466,000 deaths worldwide [114]. Brusatol inhibits growth and induces apoptosis in pancreatic cancer cells via suppression of the JNK/p38 MAPK/NF-κB/Stat3/Bcl-2 pathway [64]. Brusatol enhances the chemotherapeutic effect of gemcitabine in pancreatic cancer by inhibiting the Nrf2 signaling pathway and increasing ROS accumulation in vitro and in vivo [62]. In addition, Lu et al. have demonstrated that brusatol has potential cytotoxic effects in various types of pancreatic cancer cells, with a favorable safety profile; moreover it enhances the anticancer effects of gemcitabine and 5-fluorouracil in in vivo and in vitro models—effects associated with the suppression of the EMT process [63].
4.16 Pheochromocytoma
In the pheochromocytoma cell line hpheo1, 40 nM brusatol decreases the expression of Nrf2 protein. In the murine pheochromocytoma cell line MPC, brusatol induces cell apoptosis and decreases colony formation by elevating the levels of ROS and the accumulation of DNA oxidative damage. Furthermore, brusatol suppresses the liver metastasis of pheochromocytoma and prolongs the overall survival of tumor-bearing animals in vivo [65].
4.17 Pituitary adenoma
In the murine pituitary adenoma cell lines GH3 and MMQ, brusatol has shown potential for tumor growth inhibition and cabergoline chemosensitization in vitro and in vivo [66].
4.18 Skin cancer
In the A375 human malignant melanoma cell line, brusatol inhibits cell proliferation caused by G1 arrest and triggers cell apoptosis through downregulation of the Nrf2-mediated antioxidant response. In addition, brusatol enhances the phototherapy effect of UVA irradiation [67].
5. EFFECTS OF BRUSATOL ON VARIOUS SIGNALING PATHWAYS
5.1 MAPK signaling pathway
The mitogen-activated protein kinase (MAPK) pathway has a three-stage signaling process involving MAPK, MAPK kinase (MAPKK) and MAPKK kinase (MAPKKK), which are activated in turn and subsequently regulate cell growth, inflammation, stress, differentiation, and other important physiological and pathological effects. The MAPK signaling pathway has four main branches: ERK1/2 and ERK5, which are involved in growth and differentiation, and p38 MAPK and JNK, which are associated with growth, apoptosis and inflammation [121].
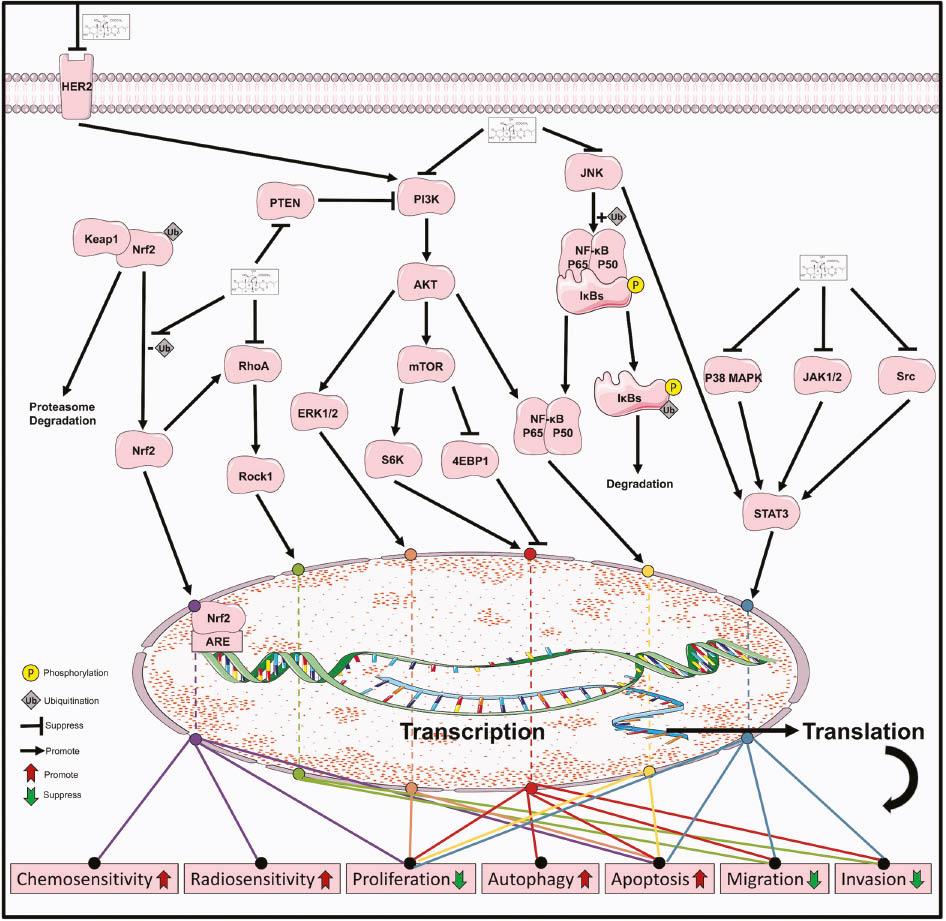
Brusatol suppresses the HER2/ERK1/2 pathway ( Figure 3 ) both in vitro and in vivo in human BT-474 breast cancer cells and SK-OV-3 ovarian cancer cells, thus enhancing the antitumor activity of trastuzumab [27]. Xiang et al. have found that brusatol inhibits growth and induces apoptosis via the JNK and p38 MAPK pathways in both PANC-1 and PATU-8988 cells, and these effects are reversed by the MAPK inhibitors SP600125 and SB203580 [64].
5.2 NF-κB signaling pathway
NF-κB is inactivated by binding the NF-κB inhibitor IκB in the cytoplasm. After the upstream signaling factors bind the membrane surface receptors, the conformation of these receptors changes, and they transmit a signal to IκB kinase, which in turn phosphorylates IκB, thus causing it to dissociate it from its complex. NF-κB then rapidly enters the nucleus from the cytoplasm, binds specific nuclear DNA sequences and promotes gene transcription. Continual activation of this pathway leads to uncontrolled growth of cells. Targeting NF-κB—such as through inhibiting the phosphorylation of IκB, the activity of NF-κB or the DNA binding activity of NF-κB—is a major topic in cancer therapy [122, 123].
Brusatol reverses lipopolysaccharide-induced EMT and induces apoptosis through the NF-κB signaling pathway ( Figure 3 ) in the gastric cancer cell line SGC-7901 [39]. Brusatol deactivates NF-κB and arrests pancreatic cancer cell growth by inhibiting Twist expression and stimulating E-cadherin expression. Moreover, Brusatol deactivates chemotherapeutic agent-induced NF-κB activation [63]. Cuendeta et al. have demonstrated that brusatol induces differentiation of the HL-60 cell line via NF-κB activation, through a process involving p50 and p65, which is inhibited by SN50, an NF-κB translocation inhibitor [47]. Xiang et al. have demonstrated that brusatol induces apoptosis and inhibits growth by inhibiting the NF-κB signaling pathway in both the PANC-1 and PATU-8988 cell lines, whereas these effects are attenuated by the MAPK inhibitors SP600125 and SB203580 [64].
5.3 PI3K/AKT/mTOR signaling pathway
mTOR, a threonine and serine protein kinase belonging to the PI3K-associated kinase family, has multiple roles in pathways involved in cell growth and proliferation [124]. mTOR is regulated by a variety of cell signaling pathways, primarily the PI3K/Akt pathway, which transmits signals through mTOR and has a major role in mediating cell survival and proliferation [125–127].
Brusatol suppresses the HER2/AKT signaling pathway ( Figure 3 ) both in vitro and in vivo in the human breast cancer BT-474 cell line and the ovarian cancer SK-OV-3 cell line, thus enhancing the antitumor activity of trastuzumab [27]. Brusatol reverses lipopolysaccharide-induced EMT and induces apoptosis through the PI3K/Akt signaling pathway in the SGC-7901 gastric cancer cell line [39]. Pei et al. have demonstrated that brusatol specifically targets the PI3K/AKT pathway in hematologic-malignancy-derived cells [45]. Ye et al. have demonstrated that brusatol exhibits anticancer effects by inhibiting cell viability and promoting autophagy-induced apoptosis via the PI3K/Akt/mTOR signaling pathway in HCC [50]. Guo et al. have demonstrated that brusatol suppresses the Akt/mTOR/S6K/4EBP1 protein synthesis signaling pathway, thus contributing to antiproliferation and proapoptotic effects in NPC [60].
5.4 JAK/STAT signaling pathway
The well-known signal transducers and activators of transcription (STAT) oncogenes are regulated by G protein-coupled receptors, receptor tyrosine kinases and interleukins. The transcription factor STAT3 dimerizes and translocates to the mitochondria or nucleus after phosphorylation, where it controls the cell cycle, cell growth, angiogenesis and cell survival—processes in which Janus tyrosine kinase (JAK), the tyrosine kinase upstream of STAT3, has a major role [128, 129].
Brusatol inhibits growth and induces apoptosis by inhibiting the Stat3 signaling pathway ( Figure 3 ) in both PANC-1 and PATU-8988 cells, whereas these effects are attenuated by the MAPK inhibitors SP600125 and SB203580 [64]. Brusatol suppresses STAT3-driven metastasis via the downregulation of EMT in HCC [49]. Brusatol inhibits the JAK/STAT3 pathway in several HNSCC cell lines, thus affecting metastasis, angiogenesis, proliferation and apoptosis [43].
5.5 Keap1/Nrf2/ARE signaling pathway
Under normal physiological conditions, Nrf2 is anchored by Keap1 in the cytoplasm. After electrophiles or ROS attack the cells, Nrf2 dissociates from Keap1, rapidly translocates into the nucleus, forms a heterodimer with Maf, binds antioxidant response elements (AREs) and then activates Nrf2-mediated expression of proteins including of antioxidant enzymes, proteins associated with the GSH redox system and proteins that act against drug resistance [130, 131].
Brusatol has been found to induce apoptosis in a non-small-cell lung cancer cell line via the ROS-mediated mitochondrial-dependent signaling pathway and Nrf2/HO-1 signaling pathway ( Figure 3 ) [21]. Brusatol suppresses the Nrf2/HO-1 pathway both in vitro and in vivo in the BT-474 breast cancer cell line and SK-OV-3 ovarian cancer cell line, thus enhancing the antitumor activity of trastuzumab [27]. Brusatol enhances the chemotherapeutic effect of gemcitabine in pancreatic cancer via the suppression of the Nrf2 pathway, which is associated with the downregulation of the antioxidant enzymes NQO1 and HO-1 [62]. The inhibitory effects of brusatol on the antioxidant enzymes HO-1 and NQO1 have been demonstrated in several studies [29, 40, 41, 46, 56]. Brusatol also interferes with the GSH redox system via the Nrf2 pathway, including reducing GSH, γ-GCS (rate-limiting enzyme for GSH biosynthesis), GCLC (catalytic subunit of γ-GCS) and GCLM (regulatory subunit of γ-GCS) [30], as also shown in other studies on brusatol [29, 40, 41, 46, 51, 65].
Aldoketo reductase family 1 member C1 (AKR1C1) is an Nrf2 target whose main function is to convert progesterone to its inactive form [132, 133]. Brusatol reverses progestin resistance by downregulating the expression of AKR1C1 and Nrf2 in endometrial cancer [38]. A similar effect of brusatol on AKR1C1, as demonstrated by Park, is associated with gefitinib resistance and EGFR-TKI cross-resistance [54].
6. TOXICOLOGICAL STUDIES
In cancer cells, brusatol exerts more powerful cytotoxicity than many current chemotherapy drugs, such as 5-fluorouracil and gemcitabine. However, in normal human GES-1 gastric epithelial cells, brusatol exerts milder cytotoxicity than gemcitabine and 5-fluorouracil. The IC50 values of brusatol/gemcitabine are 11.6 (48 h) and 9.26 (72 h), and the IC50 values of brusatol/ 5-fluorouracil are 5.58 (48 h) and 4.39 (72 h) [63].
In HBL-100 normal human breast cells, the IC50 value of brusatol is 125.5 nM (72 h) [134]. Pei et al. have demonstrated that brusatol inhibits protein synthesis with very low hematologic toxicity in normal human primary peripheral blood mononuclear cells [45]. In addition, many studies have demonstrated that brusatol suppresses brain cancer cells and decreases amyloid-β-, PFOS- and T-2 toxin-induced neurotoxicity [135–137]. However, in normal human lymphoblastoid LCL1 cells, the IC50 value of brusatol is 2.67 nM (72 h) [45].
Numerous studies have shown that brusatol at concentrations ranging from 0 to 2 mg/kg exhibits antitumor effects without significant toxicity in tumor-bearing mice [50, 60, 63]. Lee et al. have demonstrated that brusatol treatment at a high concentration of 5 or 10 mg/kg does not cause significant toxicity in experimental mice [49].
7. PHARMACOKINETICS OF BRUSATOL
The pharmacokinetics of a drug include the time course of its absorption, distribution, metabolism and excretion in the body. To investigate brusatol and further extend its applications, the in vivo activities of brusatol must be comprehensively explained. In new drug development, research on the in vivo activities of brusatol and the detection of its metabolites will offer more valuable information regarding its anticancer mechanism and safety.
The pharmacokinetics of brusatol has been examined in three articles [138–140]. Brusatol undergoes rapid absorption and distribution but is metabolized slowly after intravenous administration [138–140]. Most brusatol is excreted as metabolites, and the major metabolic pathways of brusatol in vivo have been suggested to be hydroxylation, hydrolysis and glucuronidation [138]. Regarding the distribution of brusatol in vivo, the brusatol peak concentration in the lungs is 10-fold or higher than that in other tissues (liver, kidney and spleen), thus suggesting that brusatol may actively target the lungs [139].
8. DRUG DELIVERY TOOL FOR BRUSATOL
Because of brusatol’s low water solubility and bioavailability, Chen et al. have developed a brusatol delivery system using nanoparticles modified with glycosaminoglycan–placental chondroitin sulfate A to improve its pharmacological effectiveness, thus providing an effective and safe strategy for the treatment of various tumors [141].
9. FUTURE PERSPECTIVES
Cell line misidentification problems have been known for decades, but numerous articles using the wrong cells remain published without correction. In the biomedical sciences, this ongoing problem has resulted in irreproducible experiments, false conclusions and growing concerns regarding errors. Despite decades of continued effort, this cell line misidentification problem continues [142–145]. In this article, problematic cell lines demonstrated in previous studies are denoted with “#” in Tables 1 and 2 , indicating that those articles should be interpreted with appropriate care.
In previous studies, the effects and molecular mechanisms of brusatol in many types of cancer have been reported both in vitro and in vivo. Some exceptions include bladder cancer, carcinoma of the penis, carcinoma of the vulva, esophageal carcinoma, osteosarcoma, prostate cancer and thyroid cancer. More studies are essential to demonstrate the utility of brusatol against these types of cancer and to elucidate brusatol’s mechanisms, thus decreasing the ambiguity regarding the anticancer profile of this molecule.
Despite brusatol’s promising anticancer potential, its specific targets have long been unclear. In 2011, Ren et al. [30] first identified brusatol as an Nrf2 inhibitor. Brusatol has been widely used as an Nrf2 inhibitor in many studies to date. However, the Nrf2 specificity of brusatol remains unclear. Numerous studies have used brusatol as an Nrf2 inhibitor, whereas several studies have revealed multiple mechanisms, including direct inhibition of protein synthesis [134] or downregulation of c-Myc [36, 48]. Although these hypotheses have not been confirmed through biological research, a bioinformatic method has predicted that brusatol may potentially target 464 proteins [146]. Other studies have demonstrated that brusatol specifically targets PI3K [45], STAT3 [43], SKP1 [58] and ECM1 [42]. These results indicate that many targets of brusatol may exist, and that issues of off-target effects will hinder its future applications.
Studies on the pharmacokinetics of brusatol have focused on only intravenous administration; therefore, brusatol’s behavior after intraperitoneal or oral administration remains unclear. Many studies have reported that brusatol suppresses tumor growth in vivo after intraperitoneal administration. Thus, comprehensively investigating the in vivo action of brusatol via intraperitoneal administration will be crucial to improving understanding of its anticancer mechanism. In addition, oral administration of drugs is the most convenient, economical and safe clinical treatment route. However, the pharmacokinetics and antitumor effects of brusatol in vivo via the oral route remain unclear. Evidence that brusatol shows good antitumor effects in vivo after oral administration would be highly encouraging. Furthermore, many studies have demonstrated that brusatol suppresses brain cancer cells and decreases amyloid-β-, PFOS- and T-2 toxin-induced neurotoxicity. However, whether brusatol can cross the blood–cerebrospinal fluid barrier and blood–brain barrier after these routes of administration in mice remains unclear.
Moreover, toxicological studies of brusatol remain scarce. More cytotoxicity tests are needed in normal tissue cells, together with acute and chronic toxicity tests in animals, to reveal the mechanism underlying the toxicological properties of brusatol. Simultaneously, drug toxicity indicators such as the safe dose, minimum toxic dose, maximum tolerated dose and half-lethal dose would provide a reference for subsequent preclinical and clinical trials.
Currently, novel drug delivery systems are highly promising because of their advantages, such as enhanced drug solubility, slow drug release and accurate drug delivery [147]. However, few studies have examined drug delivery systems for brusatol.
Recently, immunotherapy has become a major topic in oncotherapy. Although some articles have demonstrated that Bruceae Fructus oil can improve immunity in patients, the effects of brusatol on the immune system have not yet been studied; thus, such studies are warranted. Whether brusatol enhances immune function against different cancers and sensitizes tumors to current immunotherapies in vivo remains to be reported.
Although brusatol has shown powerful anticancer effects in multiple cancers, no clinical study has been performed, because of the lack of preclinical studies. For human welfare, researchers should conduct more preclinical and clinical studies on the pharmacological properties of brusatol, to advance understanding of brusatol and finally make this treatment clinically available.
10. CONCLUSIONS
For centuries, in efforts to combat cancer, investigators have expended enormous effort to identify more treatment strategies through various approaches. In various cancer cell models, brusatol, a primary natural constituent of Bruceae Fructus, exerts multiple effects, such as cell-cycle arrest, autophagy induction, apoptosis induction, angiogenesis suppression and metastasis inhibition, via several signaling pathways. Moreover, in in vivo animal models, its antitumor effects have been confirmed, thus indicating promising inhibition of tumor growth and prolongation of animal survival. This review described and discussed details regarding the evidence supporting brusatol’s promise as a drug candidate for cancer therapeutics.