
1. INTRODUCTION
With the quickened pace of modern life, people are faced with environmental and lifestyle changes, social factors, emotional fluctuation, and other stimuli that cause physical and mental stress. Stress causes excitation of the sympathetic-adrenal-medullary (SAM) system and hypothalamic-pituitary-adrenal (HPA) axis, and is accompanied by changes in tissue and organ function [1]. The neuroendocrine system is an important endogenous regulator of immunity. Appropriate stress is evolutionarily beneficial to life, while excessive stress has adverse effects on immunity that may accelerate the progression of neurodegenerative diseases (NDs), such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS). Currently, approximately 50 million people worldwide have NDs and the prevalence is expected to be 131 million by 2050 [2], which poses a heavy medical and public health burden worldwide; however, the mechanisms underlying NDs remain elusive.
Herpes simplex virus type 1 (HSV-1) is an enveloped double-stranded DNA virus that belongs to the Herpesviridae family. The clinical manifestations of HSV-1 include ocular infections and keratitis [3], skin, oral, and genital infections [4], and severe central nervous system (CNS) infections [4, 5]. A comprehensive epidemiologic survey of HSV-1 in Asia revealed that 50% of children and 75% of adults are infected. In addition, seroprevalence increases with age and most infections are acquired in childhood [6]. HSV-1 infection consists of four phases: acute infection; establishment of latency; maintenance of latency; and reactivation. During acute infection, HSV-1 enters epithelial cells of the peripheral mucosal tissue, then replicates in a strictly cascade manner, which ultimately leads to lytic infections. HSV-1 is a common neurotropic virus, and in addition to acute infection, HSV-1 establishes latent infections. During primary infections, the immediate early (IE), early (E), and late (L) genes are sequentially expressed to support sufficient replication in epithelial cells [7]. Following primary infection, HSV-1 from epithelial cells travels to the corresponding sensory ganglion (usually the trigeminal ganglion) by retrograde axonal transport, wherein a life-long latency is established [8]. Under a range of stimuli, including restraint stress, ultraviolet ray (UV) exposure, physical trauma, hormone imbalance, immunosuppression, or hyperthermic stress, latent HSV-1 can be reactivated and undergo productive replication. Notably, given the high infection rate in the general population, neurotropism, and lifelong persistence in neuronal cells, HSV-1 has gradually become an important factor in susceptibility to NDs.
Currently, the main drugs clinically used to treat HSV-1 infection are nucleoside antiviral analogs, such as acyclovir, which prevent the synthesis of viral DNA by inhibiting DNA polymerase [9]. Although the early efficacy of acyclovir is clear, acyclovir cannot remove the latent virus in the trigeminal ganglion or prevent the recurrence of HSV-1. Traditional Chinese medicine (TCM) has unique advantages in treating HSV-1 because of its comprehensive pharmacologic effects of multi-channel and multi-target regulation. Among the different hypotheses on the mechanism of NDs put forward in the past few years, viral infection has been suspected to be associated with pathogenesis in the progression of NDs. TCM may be used to suppress latent HSV-1 reactivation, and thus prevent and treat NDs, which may involve regulating the neuro-endocrine-immune network, treating emotional disorders, and relieving oxidative stress. This paper reviews the possible mechanisms underlying latent HSV-1 reactivation under stress and the potential effects on NDs and proposes TCM as an alternative therapy for related diseases.
2. ESSENTIAL FACTORS INVOLVED IN THE ESTABLISHMENT, MAINTENANCE, AND REACTIVATION CYCLE OF LATENT HSV-1
At present, although no natural animal host of HSV-1 has been identified, mice and rabbits are still used to study HSV-1 latent transcription, epigenetic modification, and latent reactivation [10]. After these animals are infected with HSV-1, the virus first replicates in the peripheral epithelial cells, then travels in a retrograde fashion along the axon to the sensory ganglia, after which a latent infection is established. During latency, a group of viral non-coding RNAs is expressed, known as latency-related transcripts (LATs). Although LATs promote the establishment of latency [11], LATs may not be necessary to establish latency [12]. Yu et al. [13] reported that HSV-1 can be reactivated from latency under stress conditions. HSV-1 reactivation and replication is accompanied by a decrease in LATs. Impaired IE gene expression is another prominent cause that favors the establishment of HSV-1 latency [14]. Viral protein 16 (VP16) is an essential viral envelope protein for virion morphogenesis [15]. The neuronal latency of HSV-1 may be related to the regulation of VP16 function, which may be due to its lack of or transformation from an activator to a repressor by changing the interaction of neuronal cofactors [16, 17]. The HSV-1 gene transcription cascade is initiated by VP16, which is expressed in the late stage of the virus replication cycle [18]. VP16 can promote the transcription of IE mRNAs by binding to the octamer-binding protein 1 (Oct1) in epithelial cells and host cell factor-1 (HCF-1) in epithelial and neuron cells [19, 20]. When HSV-1 is retrogradely transferred to the nucleus along the axon, VP16 enters the nucleus and combines with the Pit-Oct-Unc (POU) transcription factor family to form a triplet, which initiates the expression of IE genes [21]. Due to the absence of VP16/HCF1/Oct1 triplet, ICP0 expression is reduced, and neurons block expression of the E and L genes via the HDAC/LSD1/REST/CoREST host silencing complex, further promoting the establishment of latency.
Although many scientists are trying to unveil the mechanism underlying HSV-1 latency establishment and maintenance, no hypothesis can fully explain these life cycle events. The balance between HSV-1 lytic and latent infections is regulated by various factors, including HSV-1 encoded latency-associated transcript (LAT) [22], microRNAs [23], nerve growth factor (NGF) [1], epigenetic regulation [24], and immune response [25]. It is well-established that LAT expression is important for the maintenance of HSV-1 latency infections [26], which is related to antisense mechanisms [23], anti-apoptotic effects [27], and possible regulation of functioning protein translation [28]. Notably, in addition to LAT, other factors from the virus and the host are involved in the dynamic process of latency and reactivation of HSV-1. For example, NGF, a nerve cell growth regulator with dual biological functions (neuron nutrition and neurite outgrowth promotion), has a crucial role in the maintenance of HSV-1 latency [29]. Abundant evidence indicates that gene expression of HSV-1 is epigenetically-regulated. The changes in histone modification status control the switch between latency and reactivation. During latency, the viral genome binds to histones in the nucleus, and lytic genes are in a transcriptionally-repressed heterochromatin state. In the study of Neumann et al., the intraperitoneal administration of sodium butyrate, an inhibitor of histone deacetylase, induced HSV-1 reactivation with a frequency of 75%-100% [30]. By comparison, specific inhibitors of H3K27me3 demethylation prevented reactivation of HSV-1 in latently-infected neurons [31]. These studies indicated that the balance between latency and reactivation in the HSV-1 life cycle is exquisitely regulated by various factors.
3. ROLE OF STRESS-INDUCED ENDOCRINE ALTERATIONS AND OTHER STIMULI IN HSV-1 REACTIVATION
According to the organism response, stress is divided into physiologic, psychological, and comprehensive stress ( Table 1 ). Restraint stress in animal models has been shown to produce an inevitable physical and mental stress for induction of latent HSV-1 reactivation [45]. Our previous studies have shown that restraint stress leads to significant oxidative damage of central neurons, disruption of the endocrine system, and dysfunction of immunity, ultimately resulting in susceptibility of the host to viral infectious diseases and tumors [34, 46, 47]. In the case of HSV-1 infection, Ortiz et al. studied the modulatory effects of restraint stress on the clinical pathophysiology of primary HSV-1 infection in the SKH-1 mouse. The results showed that restraint stress prolonged viral replication during primary infection [48]. The application of restraint stress to induce HSV-1 reactivation from latency provides an experimental model to explore the complex relationships between stress and latent HSV-1 reactivation [49].
Classification of stress animal models.
| Stress type | Build method | Application | Pathophysiologic features | |
|---|---|---|---|---|
| Physiologic stress model | Cold Stress Model [32] | Put animals at low temperature (4 °C) environment | Establish acute and chronic stress models in a short time | Increase the concentration of CORT in the serum of rats |
| Heat Stress Model [33] | Place the animal in a high temperature (39 °C) environment | Research on depression and cognitive ability | Stimulate the central nerve axis to produce a series of pathophysiologic responses | |
| Restraint Stress Model [34, 35] | Fix limbs and head | Investigate the mechanism of stress damage | Induce vascular oxidative stress | |
| Fatigue Stress Model [36] | Force running and swimming | Research on drug evaluation and mechanism | Cause the organism’s immune system to decline | |
| Plantar Electric Shock Stress Model [37] | Shock plantar repeatedly | Generate a high-intensity stress response | Induce conditional fear memory | |
| Psychological stress model | Post-traumatic Stress Disorder Model [38] | Give natural enemies stimulus | Research on clinical mental disorders | Damage hippocampal function |
| Light Stress Model [39] | Change the light time to build a stress | Assess the effects of chronic stress on animals | Cause cognitive impairment | |
| Natural Enemy Stress Model [40] | Encounter natural predators or change odor | Induce acute stress response | Cause a rapid rise in serum adrenal hormone levels | |
| Restraint Stress Model [34, 35] | Fix limbs and head | Investigate the mechanism of stress damage | Induce vascular oxidative stress | |
| Mother-infant Separation Stress Model [41] | Isolate newborn mice from the mother | Assess cognitive and memory skills | Cause mental and behavioral disorders | |
| Noise Stress Model [42] | Exposure of animals to intense noise | Study diseases in susceptible people | Affect brain tissue ability to scavenge free radicals and change amino acid levels | |
| Repeated Survival Crisis Stress Model [43] | Place adventive male rats in cages of “resident” male rats | Provide a more natural stress model | Produce lasting behavioral and neurochemical changes | |
| Comprehensive stress model | Combined Physiologic and Psychological Stress Model [44] | Work together of multiple stresses | Fit the social environment comprehensively | Decrease 5-HT content in the hypothalamus |
In response to psychological stress, latent HSV-1 is reactivated from the sensory neuron, then enters the lytic cycle, subsequently causing release of the infectious virus. Because numerous hormones are involved in the stress response, it is reasonable to embrace the notion that reactivation of HSV-1 may be regulated by hormones, such as glucocorticoids (GCs) and thyroid hormones (THs) [1]. Under psychological stress, the HPA axis is activated and the concentration of GCs is rapidly increased in the blood [50]. Corticosterone (CORT) is a main type of GCs in rodents and is equivalent to cortisol in humans. CORT can establish a “long-term” stress response by binding to the glucocorticoid receptor (GR) or mineralocorticoid receptor (MR) to regulate metabolism and suppress the immune system [51]. Importantly, the elevated GCs can provoke HSV-1 replication and drive reactivation from latency.
3.1 Stress-induced elevation of GCs
Glial cells (e.g., microglia and astrocytes) have a key role in maintenance of the neuroinflammatory profile in the brain. Microglia are resident myeloid cells of the CNS that have an important role in immune surveillance [52]. Astrocytes also contribute to the immune response by releasing cytokines that influence activation of microglia and repair of neurons [53]. Overabundance of GRs in glia suggests distinct roles in inflammatory modulation. We previously reported that the stress hormone, CORT, enhances HSV-1 susceptibility in SH-SY5Y cells and a mouse model, which are largely dependent on GR. Our results further demonstrated that CORT suppresses the HSV-1-induced immune response by activating GRs, which inhibit phosphorylation of interferon regulatory factor 3 (IRF3) and expression of IFN-β, indicating the inhibitory effect of GR on anti-viral immunity [1]. There is still a lack of Reports involving the GR distribution in dopaminergic neurons or other types of neurons, however, are limited. Thus, further studies on the stress response and related diseases are warranted. The second factor involved in GR availability is transcription activity. The phosphorylation of GR has been proposed to contribute to GC resistance in multiple disease models [54]. The state of GC resistance, which may result from chronic stress [55], coincides with a decreased level of brain-derived neurotrophic factor (BDNF) in the cortex and hippocampus, and an increased level of BDNF in amygdala and dopaminergic neurons [56]. Although a majority of GR phosphorylation is glucocorticoid-dependent [57], some investigations have indicated that GR phosphorylation can be glucocorticoid-independent. For example, the BDNF-tyrosine receptor kinase B (TrkB) pathway has been reported to be involved in phosphorylation of human GRs [58, 59].
The role of GCs in HSV-1 reactivation has been extensively studied. Specifically, it has been shown that GR and Krüppel-like transcription factor 15 (KLF15) cooperatively transactivate the promoter of infected cell protein 0 (ICP0) when cells are treated with the synthetic corticosteroid, dexamethasone [60]. Additionally, Treg cells are involved in GC-induced HSV-1 reactivation. Yu et al. proposed that GCs lead to HSV-1 reactivation via increased Treg cell control of cell cluster differentiation 8+ T cells (CD8+ T cells), thus promoting viral replication under diminished immune surveillance [13]. When the HSV-1 lytic genes are expressed at a low level, CD8+ T cells secrete interferon-γ (IFN-γ) to inhibit the expression of ICP0 and prevent reactivation of the virus. Nevertheless, this process does not occur in all latently-infected neurons [1]. Moreover, intrinsic immunity is closely related to the GCs. For example, stress-induced autophagy promotes degradation of nuclear intrinsic defense components, such as promyelocytic leukemia (PML), thus mediating intrinsic immunity [61]. Promyelocytic leukemia nuclear bodies (PML-NBs) are multiprotein complexes. There is growing evidence that PML-NBs play an important role in intrinsic cellular defense against many DNA viruses, including HSV-1. Due to alternative splicing, PML has seven isoforms (I-VII), and fully functional PML-NBs requires collaboration of different PML isoforms [62]. The viral protein, ICP0, localizes to PML-NBs in HSV-1 infections, and induces proteasome degradation of PML [63]. The absence of all PML isoforms decreases cellular resistance to ICP0-null mutant HSV-1 [64]. The interaction causes the auto-lysosome degradation of PML, resulting in enhanced HSV-1 replication in neuron cells [65]. This finding is consistent with previous work in our laboratory, in which restraint stress/CORT was shown to increase HSV-1 infection by promoting the interaction of PML with autophagy-related proteins (sequestosome-1 and microtubule-associated proteins 1A/1B light chain 3B). This study provided direct evidence linking intrinsic immunity and stress-induced virus reactivation.
3.2 Stress-induced disturbance of THs
In addition to GCs, another type of endocrine hormone, TH, also has a vital role in regulating HSV-1 reactivation. Generally, TH exerts an effect via thyroid hormone receptors (TRs). The mechanism underlying HSV-1 reactivation induced by TH is distinct from that of GCs. It has been shown that TH suppresses HSV-1 replication through histone modification in differentiated neuroendocrine cells. Specifically, TH activates the transcription of LAT via TR, inhibits ICP0 expression, and recruits the repressive histones, H3K9me3 and H3K9me2, to the HSV-1 thymidine kinase promoter, thus inhibiting viral reactivation [66]. Under stimuli, such as hyperthermia and surgical trauma, TH secretions are substantially reduced and cannot effectively silence the HSV-1 gene, thus causing HSV-1 reactivation [67]. In addition, some putative TH-responsive elements (TREs) are located within HSV-1 genes, including thymidine kinase (TK) and LAT [67]. Liganded TR represses the transcription of TK in neurons to inhibit viral gene expression and promote the establishment of latency, while unliganded TR interacts with TK TREs to increase viral replication. Taken together, these results suggest that TH-mediated regulation participates in HSV-1 reactivation control.
3.3 Other stimuli
Other than endocrine alterations, UV exposure and physical trauma have been reported to be stimuli for HSV-1 latency reactivation. Rooney et al. evaluated the effect of UV light on recurrent herpes labialis in 38 patients in a double-blind, placebo-controlled crossover trial. Interestingly, following UV irradiation, herpes labialis occurred in 27 of 38 patients (71%) and the mean time to recurrence was 2.9 days. In contrast, when protected against UV using sunscreen, only 1 of 35 patients developed virus at the site of exposure [68]. Similarly, Ludema et al. proposed that UV exposure is associated with HSV recurrence. Spending ≥ 8 hours per week outdoors under high UV exposure increases the risk of ocular HSV recurrence [69].
Overall, reactivation of HSV-1 is a complex process. As a result of the host and viral interaction, it depends not only on the immune status of the host, but also on the characteristics of the virus. Therefore, the identification of regulatory factors involved in HSV-1 latency and reactivation is important for a better understanding of the underlying pathologic mechanism and clinical guidance for the treatment of related diseases. The roles of stress-induced endocrine alterations and other stimuli in HSV-1 reactivation are summarized in Figure 1 .
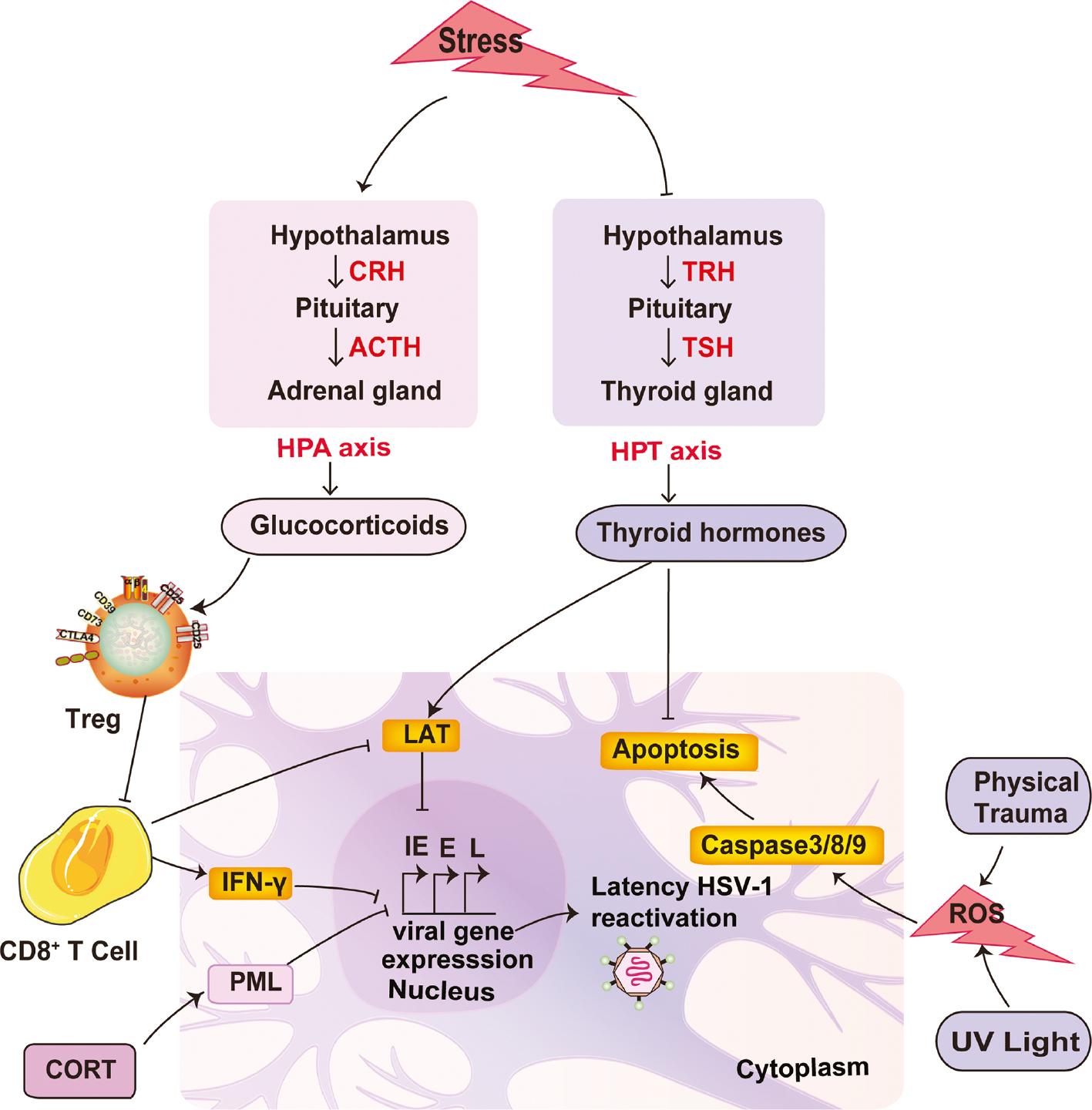
Role of stress-induced endocrine alterations and other stimuli in HSV-1 reactivation.
An important regulator of stress-induced HSV-1 reactivation is hormone levels, such as GCs and TH. Generally, restraint stress stimulates the HPA axis to increase the corticotropin-releasing hormone (CRH) released from the hypothalamus, which subsequently enhances the adrenocorticotrophic hormone (ACTH) release from the pituitary. With the increase in ACTH levels, the GCs secreted by the adrenal glands are augmented considerably. The mounting levels of GCs disrupt the host factors that are necessary for maintaining viral latency, ultimately leading to viral gene transcription and HSV-1 reactivation. The host factors impacted by GCs can be summarized as follows: 1). GCs inhibit IFN-γ secretion from CD8+ T cells in response to latently-infected neurons [1]. 2). GCs led to HSV-1 reactivation via increased Treg cell control of CD8+ T cells, thus promoting viral replication under diminished immune surveillance [13]. Other than endocrine alterations, UV exposure and physical trauma are also stimuli for reactivation of HSV-1.
4. REACTIVATION OF HSV-1 PROVOKES NEUROINFLAMMATION
Neuro-inflammation is believed to have a key role in the progression of NDs, such as AD and PD [70]. Glial cells function to support and nourish neurons cells, mainly astrocytes, microglia, and oligodendrocytes [71]. Numerous studies have shown that neuroinflammation caused by HSV-1 infection is involved in the pathogenesis of NDs [5, 72, 73]. Moreover, chronic HSV-1 infection leads to the continuous activation of microglia and triggers the production of neurotoxic and neuroprotective factors [74]. Both stress and exogenous CORT contribute to the activation and proliferation of microglia [75–77]. Once activated, microglia provide the first line of immune defense by releasing immune mediators that orchestrate innate and acquired immune responses within the CNS, including the release of pro-inflammatory cytokines [78, 79]. A study showed that activated astrocytes produce two phenotypes (A1- and A2-type) under different stimuli. A1-type astrocytes cause substantial neurotoxicity induced by three inflammatory factors (interleukin-1α [IL-1α], tumor necrosis factor [TNF], and complement component 1, q subcomponent [C1q]) secreted from microglia [80]. In contrast, A2-type astrocytes secrete various neuroprotective substances, such as glial cell-derived neurotrophic factor (GDNF), BDNF, cerebral dopamine neurotrophic factor (CDNF), and an anti-inflammatory factor (tumor growth factor-beta [TGF-β]), to maintain neuron survival and repair tissue damage during cerebral ischemia [80, 81]. Depending on the secreted cytokines, activated microglia are also classified into M1 and M2 types [82]. M1 microglia mainly secrete pro-inflammatory factors such, as tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), nitric oxide (NO), superoxide, and fatty acid metabolites [83]. In contrast, M2 microglia mainly secrete some anti-inflammatory factors, such as interleukin 10 (IL-10). These anti-inflammatory factors are inhibit the activation of M1 microglia, and promote the repair of tissues and remodeling of the extracellular matrix [84]. Exposure to stress or HSV-1 infection can disrupt the balance between pro- and anti-inflammatory responses, resulting in inflammatory mediator dominance [78, 79]. Therefore, how to maintain homeostasis between microglia and astrocytes provides new research opportunities for the treatment of neuro-inflammation.
The host recognizes HSV-1 infection through a series of germline gene-encoded pathogenic pattern receptors, such as toll-like receptors (TLRs), then produce type I interferon (IFN-I), which induces the interferon-stimulated gene (ISG) to exert an antiviral effect with the release of pro-inflammatory cytokines. A study demonstrated that HSV-1-infected microglia confer a STING-dependent antiviral state to neurons and prime IFN-I production in astrocytes via the TLR3 pathway [85]. In contrast, TLR3 is necessary to induce innate immune responses against HSV-1 in neurons and astrocytes, but not in microglia [86]. It has also been reported that Aβ-protein aggregation mimics the damage-related molecular pattern (DAMP) and promotes inflammation in microglia by activating TLR4 [87]. Although this reaction is responsible for the clearance of Aβ, the persistent pro-inflammatory condition may be harmful to the host [88, 89]. HSV-1 induces an intracranial inflammatory response through TLR2 in mice, while TLR2 knockout significantly improves the survival rate in mice [90]. It has been reported that TLR3 recognizes dsRNA [91] and induces activation of the nuclear factor-kappa B (NF-κB) signaling pathway and the subsequent release of inflammatory cytokines. In addition, inflammatory factors provoke astrocytes to produce IFN-I [92], which mediates antiviral innate immunity to fight against HSV-1 infection in the CNS.
Furthermore, studies have reported that cytokines, including IFN-γ, IL-17, and IL-6, are elevated in multiple sclerosis lesions, and C-C chemokines, including CCL2, CCL3, CCL4, CCL5, CCL7, and CXCL12, are also increased [93, 94]. Glial-derived IL-1 and IL-6 have been shown to be critical inflammatory cytokines in the brain of patients with AD [95]. In addition, cytokines, including TNF-α, TGF-β1, IL-1β, IL-2, and INF-γ, as well as reactive oxygen species (ROS)/NO are increased in the brains of patients with PD [96]. Compared with healthy individuals, the plasma levels of IL-6, IL-8, IL-4, IL-10, TNF-α, and IL-5 are significantly elevated in patients with HD. Similarly, the levels of IL-6 and IL-8 in the cerebrospinal fluid and striatum are also elevated [97]. In addition, astrocytes from patients with familial and sporadic amyotrophic lateral sclerosis are toxic to motor neurons. The toxicity is related to inflammatory molecules produced by astrocytes, including several C-C and C-X-C chemokines, TNF, and IL-8 [98, 99]. Therefore, we speculate that the expression of inflammatory factors induced by HSV-1 and immune activation state in the brain may account for increased ND susceptibility.
5. REACTIVATION OF HSV-1 INDUCES OXIDATIVE DAMAGE TO NEURONS
It has been suggested that inflammation triggered by HSV-1 reactivation may be an important cause of NDs; however, the incidence of NDs is not mitigated by suppressing neuroinflammation. Hence, there may be other mechanisms for increasing ND susceptibility. Notably, because the underlying cause of NDs is the degenerative death of neurons, it is worth determining whether HSV-1 reactivation directly damages neurons.
Oxidative stress plays a significant role in the pathogenesis of NDs because dopaminergic neurons are regularly subjected to oxidative stress, which triggers the death of neurons. Patients with PD have been shown to have reduced antioxidant capacity and lower glutathione levels [100]. Additionally, depletion of anti-oxidative substances in neurons is also related to HSV-1 reactivation. Interactions between HSV-1 and oxidative stress promote neurodegenerative processes that occur in AD. De et al. reported that antioxidant compounds prevent HSV-1 from inducing the formation of Aβ oligomers in SH-SY5Y cells, therefore suggesting that oxidative stress contributes to the pathogenesis of AD [101]. ROS induced by viral infection may defeat antioxidant defenses and cause oxidative damage to host tissues [102].
Neurons in the CNS are particularly vulnerable to oxidative stress damage compared to other organs. Exposure to oxidative stress increases the risk of neuronal damage in NDs manifested by neuronal function deficiency and neuron loss. Wang et al. showed that carbon disulfide exposure causes CNS toxicity manifested by learning and memory function impairment, which might be attributed to the extensive oxidative stress-induced apoptosis of hippocampal neurons [103]. Long-term oxidative stress in the CNS causes irreversible oxidative damage to neuron macromolecules, which leads to neuronal cell death. In fact, apoptosis, autophagy, and ferroptosis are the main pathways mediating oxidative stress-induced neuronal cell loss.
Apoptosis is activated in response to external or internal stimuli [104]. In fact, induction of host cell apoptosis is one of the consequences of HSV-1 infection, and HSV-1 induces apoptosis-related pathways at multiple stages after neuronal infection [105]. HSV-1-infected microglia produce high levels of ROS through stimulation of microglia TLR2 and the subsequent redox imbalance leads to neuronal damage, which is characterized by lipid peroxidation in murine mixed microglia neuronal cultures [106]. ROS may cause the peroxidation of cardiolipin, a mitochondrial inner membrane-specific phospholipid, and subsequent products formed by lipid peroxidation may activate intrinsic apoptosis [107]. LATs promote the survival of neuronal cells by inhibiting cell apoptosis. The anti-apoptotic function of LATs is mediated in many ways. For example, the anti-apoptotic function of LATs protects cells from cold shock-induced apoptosis by inhibiting the dephosphorylation of protein kinase B [108]. Alternatively, LATs inhibit granzyme B-induced cleavage of caspase-3 and neuronal apoptosis. In addition, LATs also prevent infected neurons from being killed by CD8+ T cells [109].
As a conserved physiologic process, autophagy has also been linked to the anti-HSV-1 effect of host. For example, autophagy removes old or damaged organelles to the cell during starvation and eliminates aggregated proteins caused by misfolding disorders to maintain cell homeostasis [110]. Under normal circumstances, ROS-induced autophagy reduces the damage caused by oxidative stress [111]; however, excessive autophagy caused by ROS leads to autophagic cell death [112]. Autophagy has a dual role in the HSV-1 infectious process. HSV-1 causes CNS system damage via autophagic regulation, which enhances neurodegeneration [113].
In addition to apoptosis and autophagy, ferroptosis, a novel iron-dependent cell death process, also participates in oxidative stress-induced neuron damage [114]. Ferroptosis is characterized by the cellular accumulation of lipid-ROS, which ultimately leads to cell death [115]. Vigorous dopamine (DA) metabolism in midbrain dopaminergic neurons is more likely to cause the release of oxidized free radicals [116]. Neurons containing various polyunsaturated fatty acids (PUFAs) are susceptible to attack by oxygen-free radicals. In addition, excessive activation of ferritinophagy induces free iron overload in cells, thus participating in the development of NDs [117]. It has been suggested that HSV-1 encephalitis in mice is associated with nucleic acid damage owing to hydroxyl radical attack, lipid peroxidation, and protein damage mediated by 4-hydroxy-2-nonenal (4-HNE) [118]. Hence, it can be inferred that ferroptosis might be involved in the oxidative damage to neurons triggered by HSV-1 reactivation, which requires further in-depth investigation ( Figure 2 ).
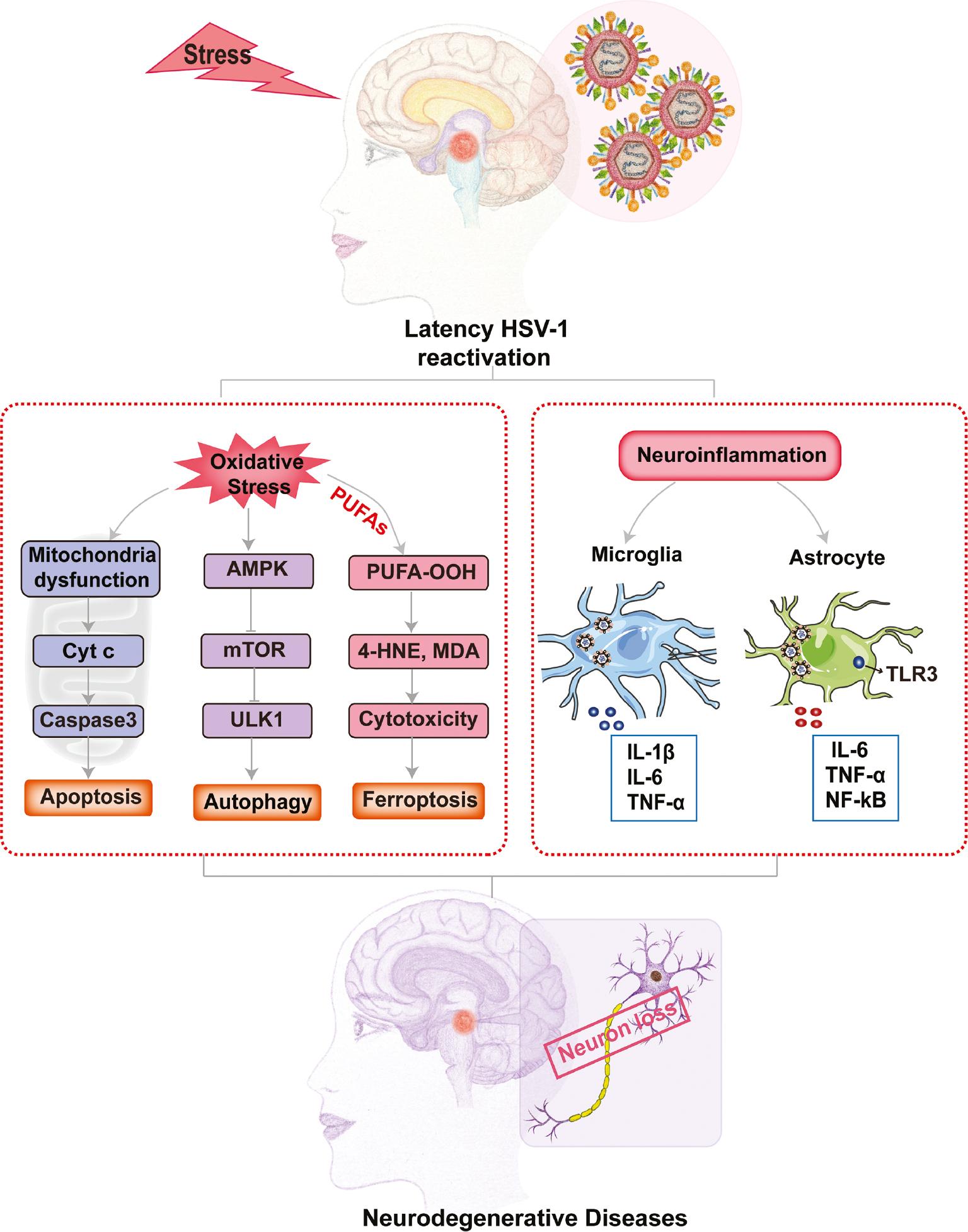
Stress-induced HSV-1 reactivation triggers neuron damage or death via ROS accumulation.
Under a series of stressors, latent HSV-1 can be reactivated. In neurons latently-infected with HSV-1, the reactivated virus induces ROS accumulation to activate the apoptosis, autophagy, and ferroptosis pathways via caspase 3 activation, oxidative stress, and lipid peroxidation, respectively, which directly leads to neural damage or death. Moreover, the reactivated virus infects microglia and astrocytes, the primary immune cells in CNS, triggering repeated pro-inflammatory cytokine secretion. Finally, the accumulating pro-inflammatory cytokines indirectly cause deleterious effects on normal or HSV-1 latent neurons.
In summary, brain tissue is susceptible to oxidative stress due to its composition and functional characteristics [119]. Oxidative stress increases with age and promotes neurodegenerative events associated with HSV-1 infection [112]. Achieving a balance between the oxidant and antioxidant systems in the brain are essential for maintaining neuronal activity and preventing neuronal degeneration [120, 121]. Based on existing research, the factors involved in maintaining a balance between oxidative and anti-oxidative stress are worthy of in-depth study and clarification. Further research will help develop neuroprotective strategies based on antioxidants, which can then be applied to the clinical prevention and treatment of NDs. Taken together, the interaction between HSV-1 and oxidative stress is important because oxidative damage occurs early in the pathogenesis of NDs.
6. REACTIVATION OF LATENT HSV-1 IS INVOLVED IN THE OCCURRENCE AND DEVELOPMENT OF NDs
NDs are characterized by progressive deterioration of CNS structure, which leads to impaired motor or cognitive function. The etiology of NDs is complex and includes genetic and environmental factors. Increased neuroinflammation and oxidative stress are key factors contributing to NDs, and in turn, oxidative damage and neuroinflammation induced by reactivation of HSV-1 may be intrinsic molecular mechanisms in the occurrence and development of NDs. In this section, we discuss the molecular signals and mechanisms in which reactivation of HSV-1 might be involved in NDs to provide new perspectives for developing therapy strategies.
6.1 Reactivation of latent HSV-1 increases susceptibility to AD
AD is a complex ND with an insidious onset and slow progression. The clinical manifestations of memory loss and progressive cognitive decline significantly reduces the quality of life. A possible link between the reactivation of latent HSV-1 and NDs has been proposed by recent studies. Wozniak et al. reported that a high frequency of HSV-1 DNA is detected in the brains of AD patients, but only a small proportion is detected in healthy control brains [122]. More recently, De Chiara et al. observed an accumulation of AD hallmarks, including amyloid-β protein, tau hyperphosphorylation, and neuroinflammation, in a murine model of serial recurrent HSV-1 infections [123]. Irreversible cognitive deficits occurred after seven cycles of reactivation [123]. Protto et al. concluded that the level of oxidative stress is increased in murine brains, and expression of 4- and 13-HNE-modified proteins were noted in the cortex after multiple stress-induced HSV-1 reactivations [124]. Taken together, HSV-1 infection is probably an important etiologic agent for AD, and the periodic reactivation of HSV-1 in the brain during episodes of stress can lead to cumulative neural cell damage [125].
6.2 Reactivation of latent HSV-1 increases susceptibility to PD
PD is a progressive ND with motor and non-motor symptoms, pathologic manifestations of dopaminergic neuron loss, and the presence of Lewy bodies in the midbrain [126]. It has been shown that apoptosis, autophagy, and ferroptosis are all involved in the degenerative loss of dopaminergic neurons. In addition, inflammatory responses and oxidative damage are also found in brains of PD patients. Primary HSV-1 infection causes neurotoxicity with increased ROS levels in the brain and neural cells [106]. It has been demonstrated that activated astrocytes are involved in central neuroinflammation, oxidative stress, and mitochondrial function abnormalities of DA neurons, which initiates death of neurons [120]. In addition, DA is oxidized by ferric ion to generate numerous reductive ions, which initiates a Fenton reaction with hydrogen peroxide. Through the Fenton reaction, hydroxyl radicals with higher cytotoxicity are generated and trigger severe lipid peroxidation, leading to apoptosis or death of substantia nigra neurons [120]. Increased ions can also cause mitochondrial dysfunction of neurons, thereby leading to dopaminergic neuron death [127]. Lipid peroxidation products, such as 4-HNE and malondialdehyde, may distort the physiologic function of neurons by crosslinking proteins and DNA, and even cause neuronal death [128]. In conclusion, oxidative stress and neuroinflammation mediated by latent HSV-1 reactivation has an important role in the pathogenesis of PD.
6.3 Reactivation of latent HSV-1 increases susceptibility to HD
HD is a hereditary autosomal dominant ND that is clinically characterized by chorea, loss of coordination, and cognitive decline with destruction of neurons in the subcortical parts of the cerebral hemispheres [129]. Oxidative stress and chronic neuroinflammation are likely complicit in driving progression of HD. It has been shown that selective activation of nuclear factor erythroid 2-related factor 2 (NRF2) signaling effectively inhibits the release of pro-inflammatory cytokines, such as IL-6, in murine microglia and astrocytes. In addition, NRF2 suppresses pro-inflammatory cytokines (IL-1, IL-6, IL-8, and TNF-α) in blood monocytes from HD patients and healthy subjects [130]. The levels of neuroinflammation-related mediator expression in the striatum, cortex, and cerebellum from post-mortem HD patient samples and controls were compared. CCL2 and IL-10 were shown to be increased in the striatum of HD patients and IL-6, IL-8, and matrix metalloproteinase 9 (MMP9) was upregulated in the cerebral cortex and cerebellum [131]. Therefore, oxidative stress and neuroinflammation mediated by latent HSV-1 reactivation have a significant role in the pathogenesis of HD.
6.4 Reactivation of latent HSV-1 increases susceptibility to ALS
ALS is the most common motor neuron disease and is characterized by the progressive degeneration of motor neurons in the brain and spinal cord [132]. To date, numerous mechanisms have been reported to be associated with ALS, including oxidative stress, neuroinflammation, excitotoxicity, impaired neurotrophic support, and mitochondrial dysfunction [133, 134]. Microglia and astrocyte proliferation were found in the motor neuron degeneration region in patients with ALS [135]. Frakes et al. reported that NF-κB is upregulated in the spinal cords of ALS patients and SOD1-G93A mice, which serves as a therapeutic target for familial and sporadic ALS patients [136]. Our previous research showed that the antioxidant enzyme, glutathione peroxidase 4 (GPX4), is significantly decreased in the lumbar spinal cord of a murine ALS model compared with the control group; however, a supplemental intrathecal injection of Gpx4-AAV significantly alleviates the symptoms and progression of ALS. In addition, Wang et al. found that GPX4 is depleted in the spinal cords of post-mortem ALS patients, which is consistent with the spinal cord and early brain findings in transgenic animal models with ALS [137]. Oxidative stress and neuroinflammatory responses may be responsible for the ALS pathologic process. Therefore, we reasoned that latent HSV-1 reactivation exacerbates disease progression of ALS by inducing neuroinflammation and oxidative damage of motor neurons.
7. POTENTIAL THERAPEUTIC EFFECT OF TCM ON HSV-1-INDUCED NDS
TCM is an effective clinical resource for the treatment of NDs, but little evidence indicates the relationship between the neuroprotective effect of TCM and inhibition of latent HSV-1 reactivation. Currently, no treatment eliminates latent HSV-1 virus in neurons, while acyclovir and other purine nucleoside analogues only inhibit the replication of reactivated virus with increasing drug resistance. An advantage of TCM is prevention of latent HSV-1 reactivation and increasing the difficulty of virus reactivation by regulating the host NEI network. In addition, the antioxidative and antiviral effects of TCM also controls viral replication and oxidative stress in host cells, thus reducing damage to central and peripheral tissues ( Figure 3 ). Hence, we discuss the possible neuroprotective mechanism underlying TCM through inhibition of HSV-1 reactivation and replication in four ways.
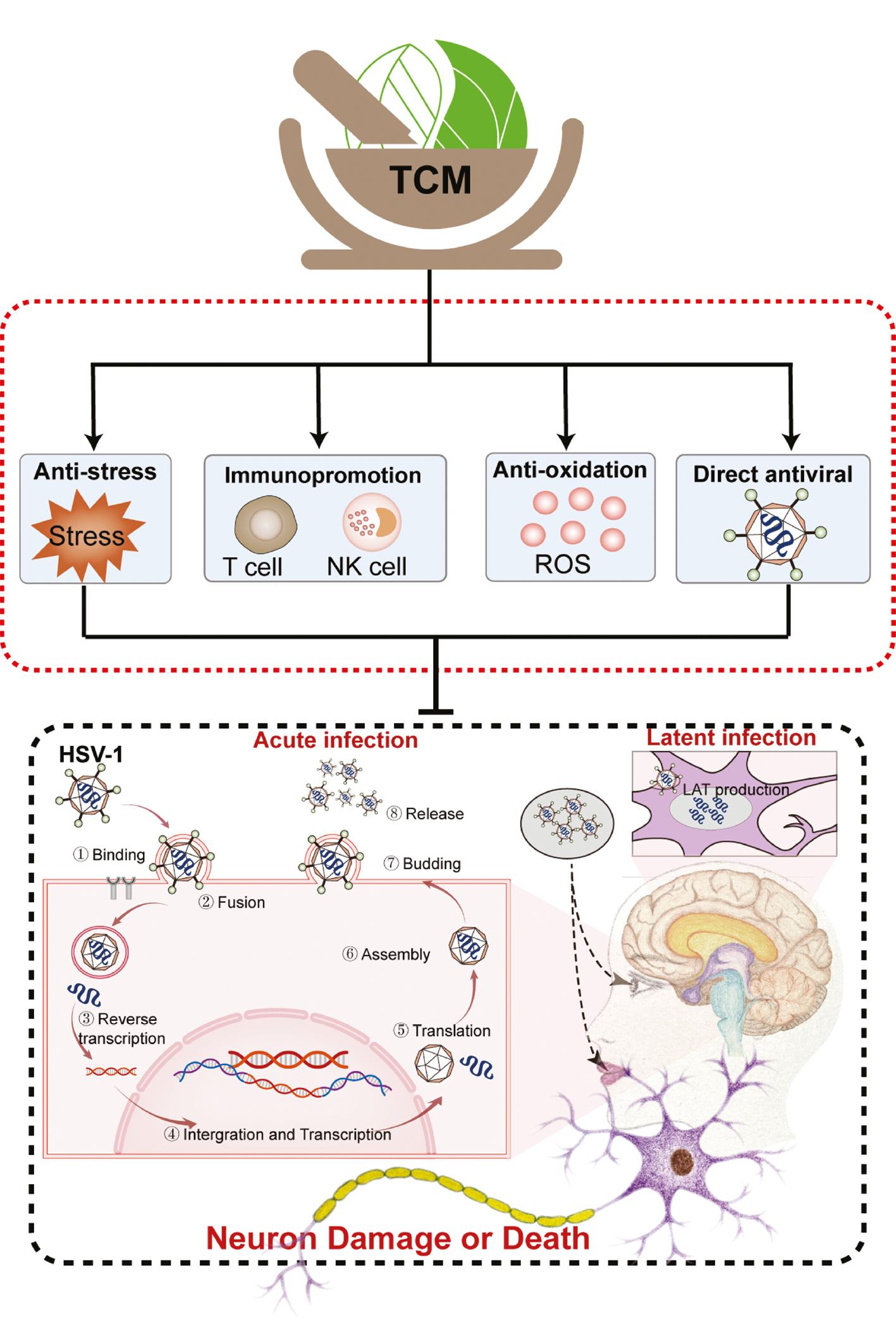
Potential therapeutic effects of TCM on HSV-1-induced NDs.
TCM alleviates HSV-1-induced NDs by controlling viral replication and reactivation and reducing damage to neurons. The inhibitory mechanism underlying TCM against HSV-1 activation and replication can be summarized as follows: 1) anti-stress effects; 2) immunomodulatory effects; 3) anti-oxidant effects; and 4) anti-viral effects.
7.1 Anti-stress TCM activity
TCM is widely used for the treatment of a variety of physical and psychological stress-related disorders in China. The anti-stress TCM activity is related to a regulatory effect on the neuroendocrine system. Collective evidence supports the inhibitory effect of TCM on the overactive HPA axis and abnormal GC release under stress conditions [138, 139]. Our previous studies showed that excessive GCs impair host innate immunity and render the host susceptible to influenza viruses and HSV-1 [65, 140]. As mentioned above, GCs inhibit host immune surveillance of latent HSV-1. TCM may control GC levels in stressed hosts, and hence reduce latent HSV-1 reactivation and the risk of non-hereditary NDs. In addition to Chinese medicine, TCM includes non-drug therapies, such as acupuncture, that has been shown to modulate the NEI network according to recent studies [141]. Similarly, electroacupuncture, an improved form of traditional Chinese acupuncture, inhibits hyperactivity of the HPA axis for the treatment of depression or depressive disorder [142]. Electroacupuncture has been shown to improve HSV-1 infection-induced facial nerve palsy with a decrease in HSV-1 DNA content in mice, indicating the efficacy of electroacupuncture to promote the recovery of nerves from HSV-1 infection-related injury [143]. Harmine, isolated from the seeds of Peganum harmala, is a natural tricyclic β-carboline alkaloid with a core indole structure and a pyridine ring [144]. Harmine is a selective inhibitor of tau phosphorylation at multiple AD-related sites. Frost et al. showed that β-carboline compounds, including harmine, not only effectively reduces the expression of phosphorylated forms of tau protein, but also inhibits the direct phosphorylation of tau protein on serine 396 catalyzed by the DYRK1A gene [145]. Harmine blocks HSV-1 infection by downregulating the cellular NF-κB and MAPK pathways [146]. In 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD mice, baicalin treatment effectively prevents the loss of TH-positive neurons in the substantia nigra and decreases the DA content in the striatum [147]. Our previous results indicated that the anti-HSV-1 effect of baicalin involves two pathways, including inactivation of viral particles and inhibition of phosphorylation of IκB kinase-β (IKK-β) [148]. Baicalin is a promising candidate for the treatment of HSV-1 infection and reactivation.
7.2 Immunomodulatory effect of TCM
TCM regulates host immune function to cope with exogenous pathogenic microbe infections. Three TCM decoctions and three formulated Chinese medicines have been widely applied in the treatment of COVID-19 in the absence of effective anti-SARS-CoV-2 drugs in China [149, 150]. It has been reported that TCM and its active ingredients modulate innate and acquired immunity. In addition to exerting an effect on the neuroendocrine pathway, TCM also has a direct regulatory effect on immune cells [151, 152]. Several studies have demonstrated that TCM enhances immune defenses of the host against HSV-1. Shi et al. discovered that astragalus polysaccharide promotes TLR3/NF-κB-mediated immunologic response and hence protects astrocytes from HSV-1 infection-induced cell damage [153]. Total flavonoids from Ixeris Sonchifolia markedly increase the levels of IL-2 and INF-γ in serum of HSV-1 infected mice, and alleviates pathologic damage to the cornea [154]. Moreover, moxibustion, another non-pharmaceutical technique in TCM, markedly increases the survival rate of mice infected with lethal doses of HSV-1, and is correlated with the augmentation of natural killer cell activity and cytokine production, such as IFN [155]. Therefore, TCM might reduce damage to the CNS through inhibiting latent HSV-1 reactivation or improving the clearance of HSV-1 by immunity enhancement of people under stress or with low immunity.
More recently it has been shown that gut microbiota and its metabolites act as an important regulator of the neuroimmune system, and a dysregulated gut microbiome-neuroimmune axis contributes to neurologic disorders [156]. A recent study showed that an intestinal neomycin-sensitive bacteria metabolite, nicotinamide N-oxide, inhibits microglia activation and neuroinflammation of the CNS by promoting mitophagy to prevent herpes simplex encephalitis [156]. Polysaccharides are common active ingredients in TCM, which can be degraded and absorbed by intestinal bacteria for production of short-chain fatty acids (SCFAs), which are beneficial to host hemostasis [157]. Importantly, SCFAs are recognized as a mediator in gut-brain axis communication to help the recovery of various neurologic diseases, including AD and PD [158]. In addition, TCM polysaccharides modulate the composition of the beneficial and pathogenic bacteria to relieve intestinal dysbiosis due to an unbalanced diet, antibiotic treatment, stress stimuli, or some disease conditions [159].
7.3 TCM antioxidative effect
Chinese medicinal herbs are a source of natural antioxidants against oxidative stress or oxidative damage and show great potential for neuroprotection of dopaminergic neurons in the treatment of PD [160, 161]. In fact, counteracting oxidative stress is an important strategy to prevent or treat a wide variety of NDs. Free radical scavenging can reduce oxidation of protein, DNA, and lipids, and inhibit downstream signaling pathways activated by ROS to protect neurons from inflammatory autophagy and cell death [162]. Furthermore, Kim et al. found that a Mori ramulus ethanol extract and its component (morusin) block HSV-1-induced production of ROS in Vero cells, which may explain the antiviral activity [163]. The relationship between oxidative stress and HSV-1 replication, however, needs to be further elucidated. The neuroprotective effect of TCM based on mechanisms against oxidative damage is an important approach to prevent and treat neurodegenerative diseases.
7.4 TCM antiviral effect
The damage of host cells induced by latent HSV-1 reactivation also depends on the outcome of viral replication. Hence, TCM functions as an antiviral reagent to inhibit viral replication in neurons, reducing activation of microglia and subsequent neuroinflammation. We have thoroughly reviewed the anti-HSV-1 effects of TCM and its active ingredients in vitro and in vivo [164]. In addition, our laboratory found that baicalein, a flavonoid from Scutellaria baicalensis Georgi, demonstrates a strong inhibitory effect on the replication of HSV-1/F and acyclovir-resistant HSV-1/Blue through dual mechanisms of inactivating viral particles and inhibiting the phosphorylation of IKK-β [148]. Hence, TCM not only directly inhibits the activity of virus, but also regulates the function of host cells to reduce their susceptibility, which may explain the decreased resistance that has emerged in the treatment of viral infectious diseases with TCM.
8. CONCLUSIONS AND PERSPECTIVES
With acceleration of the life rhythm, the increasing work pressure and environment deterioration has become an extremely important factor in the occurrence, development, and recovery of diseases. TCM has proposed the theory of emotion changes inducing diseases, and it is gaining increasing value and significance in the etiology of modern diseases. At present, HSV-1 is one of the most widespread viruses known to cause acute and recurrent infections in humans, with an adult infection rate of 60%–95% [165]. The stress of the organism is the direct cause of HSV-1 recurrence, and the occurrence and development of NDs are closely related to latent pathogens in the brain. The incidence of dementia among individuals infected with HSV-1 is 2.564 times the control group, and the use of anti-herpes drugs is associated with a reduced risk of dementia [166]. When exposed to stress events, latent HSV-1 is repeatedly reactivated, leading to nerve cell damage and accelerating the progression of NDs [123, 167]. Therefore, improving an organism’s ability to resist stress or reduce susceptibility to recurrence caused by stress will help prevent HSV-1 reactivation. In this review we discussed stress-induced reactivation of latent HSV-1, which not only induces an organism’s neuroinflammatory reaction, but also leads to an oxidative stress response. The mechanism by which stress causes HSV-1 reactivation needs further exploration. In addition, we also discussed the anti-HSV-1 mechanism underlying TCM from multiple components, targets, and perspectives. It has been demonstrated that TCM and its active ingredients have broad prospects in the treatment of HSV-1.