
Introduction
Most research on the cardiovascular system has focused on its blood circulation function. It was rarely considered to be related to inflammation and immunity. The origin of cardiovascular immunology research may date back to 30 years ago. The first autoantibody targeting the myocardial molecule adenine nucleotide translocator was found by Schultheiss and Bolte [1] in 1985; it acts against the adenine nucleotide translocator of myocardial mitochondria in patients with dilated cardiomyopathy (DCM). One after another, more autoantibodies were defined, including the autoantibodies against β1-adrenergic receptor, myosin heavy chain, M2 cholinergic receptor, and L-type calcium channel in patients with DCM and viral myocarditis. The pathogenesis of chronic heart failure (CHF) was mainly considered to be the imbalance of hemodynamics and neuroendocrine function until 1990. Then Levine et al. [2] found that circulating levels of tumor necrosis factor (TNF) are related to the aggravation of CHF. It was more interesting that autoantibodies were found in patients with hypertension. Fu et al. [3] found autoantibodies against α1-adrenoceptor in patients with malignant hypertension in 1994. Autoantibodies against α1-adrenoceptor and angiotensin II receptor type 1 (AT1 receptor) were found in patients with preeclampsia and refractory hypertension [4]. In 1999, Ross [5] proposed the theory that atherosclerosis is a kind of inflammatory disease. Therefore cardiovascular immunology research is accompanied by investigation of the autoantibodies, cytokines, and inflammatory cells in cardiovascular diseases such as DCM, hypertension, atherosclerosis, acute myocardial infarction (AMI), and CHF. This cardiovascular immunology study was conducted in Wuhan Union Hospital from 1991, and it mainly covered (1) the autoantibodies associated with cardiomyopathy and hypertension, (2) T-cell subsets and cytokines related to heart failure, and (3) a therapeutic vaccine for the prevention and treatment of hypertension.
Autoantibodies Associated with Cardiomyopathy and Hypertension
The molecular mimicry analysis of viral proteins with myocardial proteins, applied by Hoebeke [6], showed high homology in 1996, which prompted the study of anti-myocardial antibodies in patients with acute viral myocarditis and DCM. The molecular mimicry theory greatly drove the progress of cardiovascular immunology research. Liao et al. [7–9] began to report anti-myocardial antibodies in DCM patients and their pathogenic mechanisms from 1993. Yuan et al. [10] found that Th17 cells are hyperactive in patients with acute viral myocarditis, assisting B cells to produce anti-heart adenine nucleotide translocator antibodies, while Th2 cells facilitate the activation of B cells in DCM. Sudden death is very common in patients with DCM. This was suspected to be related to some antibodies from DCM. Xiao et al. [11] found antibodies against L-type calcium channel in sera of patients with DCM in 2011 (see Table 1). The affinity purified autoantibody against L-type calcium channel induces ventricular arrhythmias and action potential changes, prolongation of action potential duration and early afterdepolarization, and causes ventricular tachycardia and sudden death.
Antibodies Against L-type Calcium Channel Induce Ventricular Arrhythmias and Sudden Death in Dilated Cardiomyopathy Patients.
| CC-AAbs positive (n=39) | CC-AAbs negative (n=41) | P-value | |
|---|---|---|---|
| Age (years) | 53±14 | 51±15 | NS |
| Sex, female/male | 9/30 | 9/32 | NS |
| Presence of ventricular arrhythmias | 36/39 (92.3%) | 29/41 (70.7%) | 0.013 |
| VPB | 35/39 (89.7%) | 27/41 (65.9%) | 0.011 |
| VPB (frequency in 24 h) | 3214±2587 | 1409±2165 | <0.001 |
| VT | 23/39 (59.0%) | 10/41 (24.4%) | 0.002 |
| VT (frequency in 24 h) | 48±51 | 41±39 | 0.035 |
| Follow-up (32±8 months) | |||
| Primary end point (sudden death) | 8/39 (20.5%) | 2/41 (4.9%) | 0.045 |
| Secondary end point (all-cause death) | 10/39 (25.6%) | 8/41 (19.5%) | NS |
CC-AAbs, antibodies against L-type calcium channel, NS, not significant; VPB, ventricular premature beat; VT, ventricular tachycardia.
DCM is often a fatal cause of heart failure. It has recently been confirmed that the pathogenesis of DCM is related to autoimmune reaction. DCM usually worsens gradually. Some patients are asymptomatic; however, their left ventricle may have been dilated for months or even years. Myocardial damage mediated by autoantibodies is the main cause of DCM, and the damage may be inhibited by a calcium antagonist. The Intervention Study of Diltiazem in Dilated Cardiomyopathy (ISDDC), a multicenter, randomized and placebo-controlled clinical trial in 16 hospitals of China was reported by Liao [12] in 1998. From the analysis of the clinical findings of ISDDC, it was concluded that the calcium antagonist diltiazem significantly reduces mortality and the heart failure hospitalization rate in patients with DCM (see Table 2). The purpose of the trial was to study the clinical changes with DCM stage and the effects of intervention at different stages of DCM (see Figure 1).

Immunopathogenesis of and Immunotherapy for Dilated Cardiomyopathy (DCM).Ab, antibody; HF, heart failure; anti-β1-RAb, anti-β1-adrenergic receptor antibody.
Prognosis Analysis of Dilated Cardiomyopathy Patients in the Intervention Study of Diltiazem in Dilated Cardiomyopathy.
| Placebo control (n=107) | Diltiazem (n=114) | |
|---|---|---|
| Ambulatory treatment | 63 (58.9%) | 102 (89.5%)*** |
| Repeated hospitalization | 44 (41.1%) | 12 (10.5%)*** |
| Death | 12 (11.2%) | 4 (3.5%)** |
Compared with placebo group, **P<0.05, ***P<0.01.
Higher levels of autoantibodies against AT1 receptor and α1-adrenoceptor in patients with hypertension were detected, particularly in those with refractory hypertension, in 2002. Meanwhile, abnormal activation of the neural-humoral-immune system in refractory hypertension was found [13]. The autoantibodies were confirmed to cause cardiac and arterial remodeling [14, 15], and they were associated with the increased recurrence of stroke and death from stroke in hypertensive stroke patients [16, 17]. A multicenter, randomized, parallel-group comparison clinical trial, Study of Optimal Treatment in Hypertensive Patients with Anti-AT1-Receptor Autoantibody (SOT-AT1), was conducted in 2011, with regimens based on candesartan (an angiotension receptor blocker) or imidapril (an angiotensin-converting enzyme inhibitor). From the clinical trial it was confirmed that the antihypertensive effect of the angiotension receptor blocker is better than that of the angiotensin-converting enzyme inhibitor, suggesting that anti-AT1 receptor autoantibodies can induce hypertension [18] (see Figure 2). SOT-AT1 provided a basis for targeted therapy of hypertension.
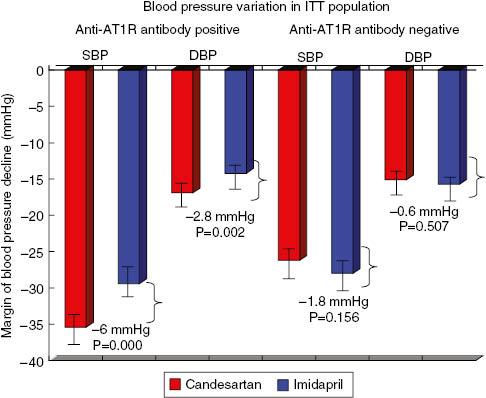
Blood Pressure Variation in the Intent-to-Treat (ITT) Population According to Anti-Angiotensin II Receptor Type 1 (anti-AT1R) Antibody Stratification Analysis.Data were adjusted for the baseline values. The mean difference is significant at the 0.025 level. The ITT population included all patients who were randomly assigned to receive the study drugs. DBP, diastolic blood pressure; SBP, systolic blood pressure.
T-Cell Subsets and Cytokines Related to Heart Failure
The relationship between circulating levels of TNF and heart failure was worth studying. Lymphocyte infiltration was observed in the infarct area of AMI rats in 2002 [19]. It can also be present in the myocardial interstitium of normal rats after adoptive transfer. Anti–myosin heavy chain antibodies can be detected in both AMI rats and rats that have undergone transfer [20]. This suggested that the immune system is activated after AMI.
The anti-TNF therapy against congestive heart failure (ATTACH) trial indicated that the TNF-α monoclonal antibody infliximab increased mortality and the hospitalization rate for heart failure deterioration in 2003 [21]. Cheng et al. [22] found that the level of TNF-α was elevated markedly in cardiomyocytes of AMI rats in 2005. This observation suggested that infliximab targets TNF-α on the myocardial membrane. However, they did not determine whether the TNF-α expressed in cardiomyocytes was from the circulation or myocardium; therefore they hypothesized the autocrine effects of TNF-α after AMI. In 2012, Yu et al. [23] showed that hypoxia promotes the stable expression of hypoxia-inducible factor 1α and its transfer into the nucleus. Hypoxia-inducible factor 1α combined with HRE-8, a hypoxia response element in the TNF-α gene promoter region, to initiate the transcription of TNF-α. At the translational level, TNF-α protein occurs in two forms. It is initially expressed as a 26-kDa transmembrane protein (mTNF), and after cleavage by a TNF-converting enzyme, a 17-kDa soluble protein (sTNF) is released. mTNF was found the main form of TNF-α released from hypoxic cardiomyocytes. It was carried out of hypoxic cardiomyocytes by exosomes, and then transported to target cells to induce the apoptosis of themselves or other cardiomyocytes. This study clarified the mechanism of TNF-α autocrine effects after AMI (see Figure 3). In 2013, Yu et al. [24] firstly suggested that TNF-α-secreting B cells contribute to myocardial fibrosis, which was a new DCM pathogenesis mechanism.
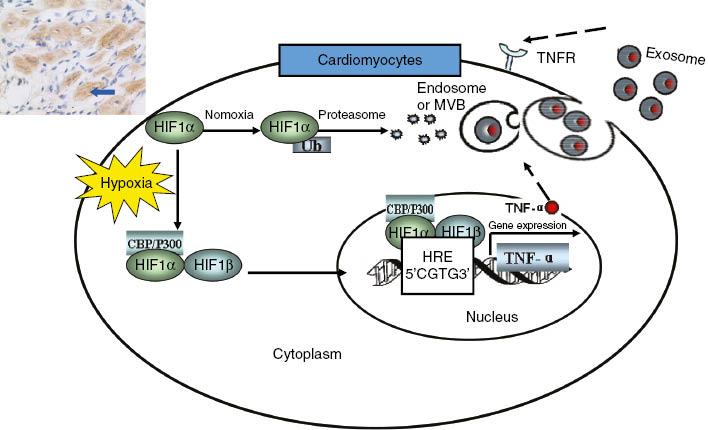
Mechanisms of Tumor Necrosis Factor α (TNF-α) Autocrine Effects in Acute Myocardial Ischemia, Hypoxia Induces Accumulation and Expression of Hypoxia-Inducible Factor 1α (HIF-1α) in Cardiomyocytes.HIF-1α initiates transcriptional activation of TNF-α in response to hypoxia. mTNF is secreted by hypoxic cardiomyocytes and released through the exosomal pathway. CBP, cyclic AMP response element binding protein binding protein; HIF-1β, hypoxia-inducible factor 1β; HRE, hypoxia response element; MVB, multivesicular body; TNFR, tumor necrosis factor receptor; Ub, ubiquitin.
Cheng et al. [25] found that the functional imbalance of T-cell subsets mediates myocardial injury and CHF, and showed the hyperfunction of Th1/Th17 in AMI and acute viral myocarditis through clinical and experimental studies. They found elevation of local IL-17A levels in myocardial ischemia–reperfusion (I/R)-injured cardiomyocytes mainly derives from γδ T cells and that IL-17A accelerates cardiac function harm of I/R-injured mice [26]. In CHF patients, the number of circulating T regulatory cells (Tregs) decreased, as did their inhibitory effects. Tang et al. [27–29] confirmed the causes of Treg defects, including thymic dysfunction, increased apoptosis of Tregs, and responsiveness decrease of responder T cells. Adoptively transferred Tregs alleviated myocardial inflammation after AMI; the mechanism included the myocardial inflammation restrained by adoptively transferred Tregs and the apoptosis and fibrosis of cardiomyocytes [30]. It was found that statins, beta blockers, and qiliqiangxin can reduce the proportion of Th1 cells and increase the proportion of Tregs. In addition, renin-angiotensin system inhibitors such as captopril, irbesartan, and olmesartan also reduced TNF-α, IL-1, and IL-6 expression in the hearts of AMI rats, and they can also significantly improve survival, cardiac function, and ventricular remodeling after AMI [31–33]. These cardiovascular drugs can also regulate the imbalance of the expression of the inflammatory cytokines TNF-α and IL-10 in lymphocytes and cardiomyocytes, and prevent ventricular remodeling [22, 34–36]. O the basis of the results of the clinical and experimental studies mentioned above, we proposed the immunopathogenesis of heart failure, which provided a new theoretical basis for immunoregulation therapy for heart failure (see Figure 4).
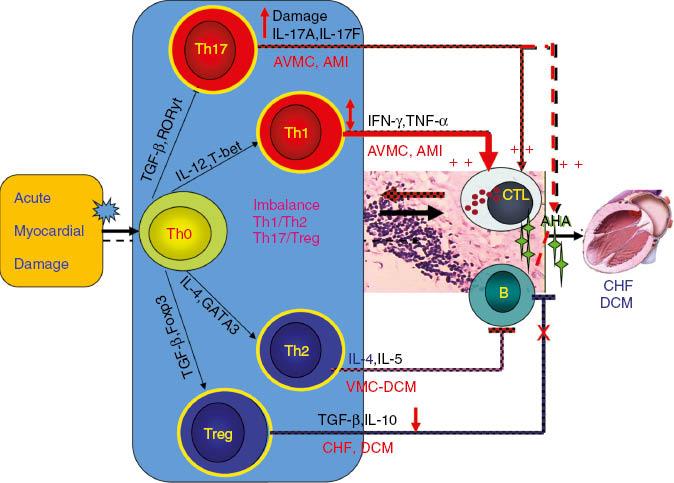
Mechanism of T-Cell Subset Imbalance After Acute Myocardial Damage.AHA, anti-heart antibodies; AMI, acute myocardial infarction; AVMC, acute viral myocarditis; CHF, chronic heart failure; CTL, cytotoxic T cell; DCM, dilated cardiomyopathy; IFN, interferon; TGF, transforming growth factor; TNF, tumor necrosis factor; VMC, viral myocarditis.
Therapeutic Vaccine in Prevention and Treatment of Hypertension
Vaccines have the characteristics of precision and long-term effect, and may be used in the treatment of chronic diseases. Zhu at al. [37] and Li et al. [38] invented the first antihypertensive polypeptide vaccine ATR12181 against AT1 receptor in 2005 [37, 38]. In 2010, Chen et al. [39] constructed the technical platform for polypeptide vaccines and the improved ATRQβ-001 antihypertensive vaccine (see Figure 5). ATRQβ-001 vaccine could not only effectively reduce blood pressure and protect target organs but could also significantly inhibit the progression of atherosclerotic plaques and reduce renal pathological damage in diabetic nephropathy rats [39–41]. ATRQβ-001 vaccine is different from angiotensin receptor blockers in the mechanism of action. It does not activate the renin-angiotensin system by a feedback loop. It was verified to downregulate the angiotensin-converting enzyme–angiotensin II–AT1 receptor axis and upregulate angiotensin I-converting enzyme 2–angiotensin 1-7–Mas receptor axis at the same time [41]. Therefore, the ATRQβ-001 antihypertensive vaccine is a novel renin-angiotensin system regulator. ATRQβ-001 vaccine should be injected subcutaneously once a month, making it convenient to manage systematically. A successful transnational study of antihypertensive vaccines will improve adherence and increase the control rate in the long-term treatment of cardiovascular diseases. It will also improve the precise management of chronic diseases.
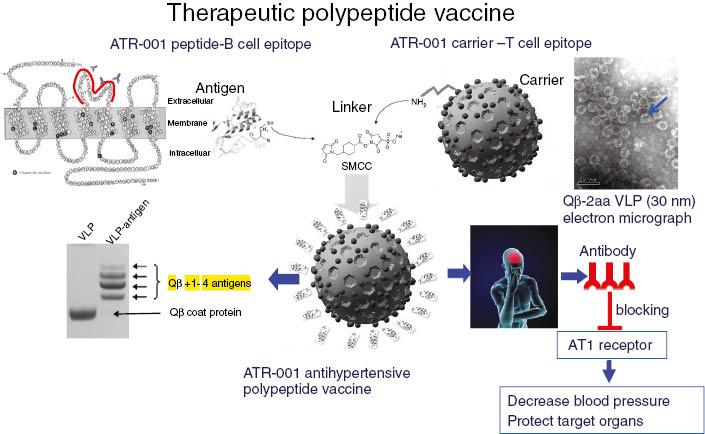
Construction and Antihypertensive Effect of the ATRQβ-001 Therapeutic Polypeptide Vaccine.The ATR-001 peptide is covalently conjugated to virus-like particles (VLPs) with use of a sulfosuccinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sulfo-SMCC) linker to produce the ATRQβ-001 vaccine. Anti-ATR-001 antibodies specifically bind to angiotensin II receptor type 1 (AT1 receptor) and inhibit its activation by angiotensin II. ATRQβ-001 vaccination effectively decreases blood pressure and protects target organs from hypertension-induced damage.
Conclusion
Inflammation and immunity play an important role in the pathogenesis of DCM, hypertension, atherosclerosis, AMI, and CHF. Autoantibodies, cytokines, and T-cell subsets play a major role in mediating cardiovascular diseases. In the past 25 years, cardiovascular immunology research in Wuhan Union Hospital found autoantibodies against L-type calcium channel that result in ventricular tachycardia and sudden death in DCM, explained the mechanism of functional imbalance of T-cell subsets and TNF-α autocrine effects in AMI, and invented the first antihypertensive polypeptide vaccine (ATRQβ-001). The book Cardiovascular Immunology was published in 2008 and introduced the basic theories of cardiovascular immunology, immunopathogenesis, and targeted therapy of clinical diseases, and experimental methods of cardiovascular diseases research [42]. In the future, there will be more cardiovascular immunology research in Wuhan Union Hospital.