
Introduction
Long QT syndrome (LQTS), a common inherited cardiac disorder caused by mutations in genes encoding cardiac ion channels, is characterized by prolonged QT intervals detected on the surface ECG.
Through disruption of the normal action potential process, LQTS, which often results in severe forms of arrhythmia such as ventricular fibrillation, is associated with clinical manifestations including syncope, palpitations, and sudden cardiac death mainly in the pediatric age group [1, 2].
With ever-expanding knowledge of the genetic and clinical profiles of LQTS, LQTS has been diagnosed in more and more patients. According to one study, the estimated prevalence of LQTS is believed to be in the range from 1 in 2000 to 1 in 2500 [3]. Since the discovery of an LQTS-causing gene in 1995 by means of sequencing technology, a plethora of mutations in 15 genes have been found [4]. Among these, KCNQ1, KCNH2, and SCN5A are the top three and account for nearly 80% of the genotyped LQTS patients [5]. A single nucleotide replacement in one of the culprit genes that results in substitution of an amino acid accounts for the vast majority of known mutations. However, multiple mutations, usually more than two mutations in the same gene or in different genes, are relatively rare. Here we present digenic mutation discovered in an LQTS-affected Chinese family through whole-exome sequencing. The data show how this complicated mutation can have an impact on the phenotype of the affected members.
Materials and Methods
This study protocol was approved by the Ethical Committee of the First Affiliated Hospital of Zheng Zhou University in Henan province, China, and was implemented strictly in accordance with the Declaration of Helsinki. All participants were well informed and all gave their written consent.
Patient and Other Participants
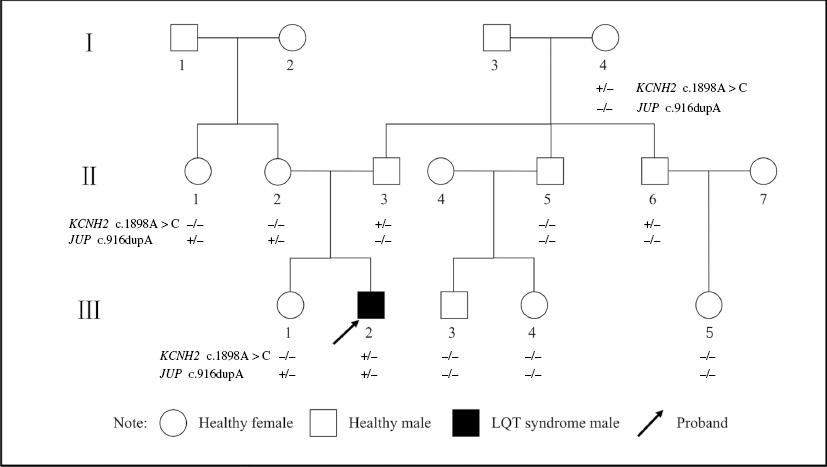
A family of three generations with 11 members (Figure 1) from Henan province in central China participated in this study. The proband, a 15-year-old boy who had experienced sudden happened syncope and seizure many times during the 5 years since the symptoms first occurred, was transferred to our hospital when he experienced convulsion and loss of consciousness, which suddenly occurred after exercise training and were not relieved by medical treatment at the local hospital. Both his father and his aunt have a history of syncope, while the rest of the family members do not report similar conditions.
Genetic Screening
Genomic DNA was extracted from anticoagulant (EDTA)-containing blood drawn from each participant’s peripheral vein by a blood DNA extraction kit from QIAGEN (Beijing). A complementary DNA (cDNA) library was built and a capture assay was performed in accordance with the instructions for the Agilent SureSelect Human All Exon kit. Only the qualified cDNA library went through deep and high-throughput sequencing on the Illumina HiSeq platform. Sequencing results were aligned with the hg19 reference sequence to find any potential variations. Then site-specific validation was performed by Sanger sequencing for the proband with use of specially designed primers targeting potential variation regions of 46 inherited cardiac arrhythmia–related genes. The genes are listed in Supplementary Table 1. After detection of the causative mutation from the proband, Sanger sequencing was performed for the family members to confirm co-segregation effects in the family. The primer sequences are given in Supplementary Table 2.
Results
Clinical Features of the Proband
The 15-year-old-boy often struggled with lethal arrhythmias and randomly occurring short periods of unconsciousness. After his arrival at our hospital, a detailed medical history was promptly obtained and a detailed physical examination was promptly performed.
According to his medical history, coupled with limb convulsion, tightly closed teeth, urinary incontinence, and cyanosis of the lip, loss of consciousness, lasting nearly 5 minutes, suddenly occurred after regular school military training. Similar symptoms recurred with greater frequency and lasted longer than before. Sudden cardiac arrest occurred three times during his stay in the local hospital and he was successfully resuscitated by the prompt action of the physicians. According to a local physician’s statement, the patient’s ECG revealed a short period of torsades de pointes. Because of poor respiratory function, oral tracheal intubation was applied. The day after intubation, ventricular fibrillation occurred. Luckily the heart rhythm returned to sinus rhythm after electric defibrillation.
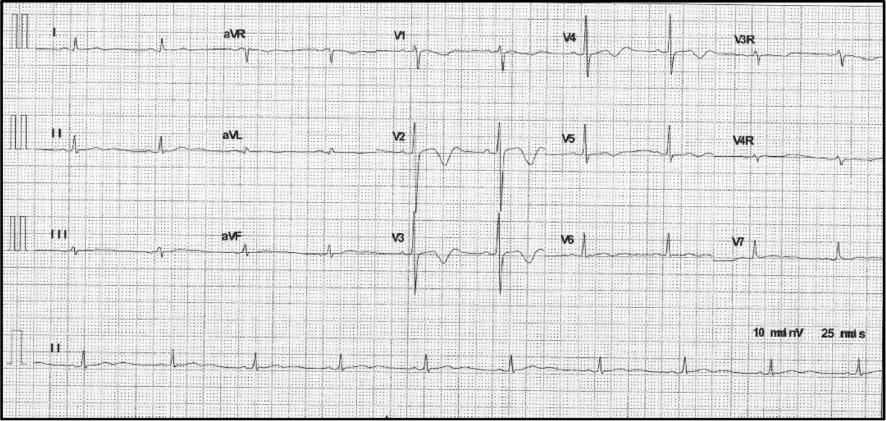
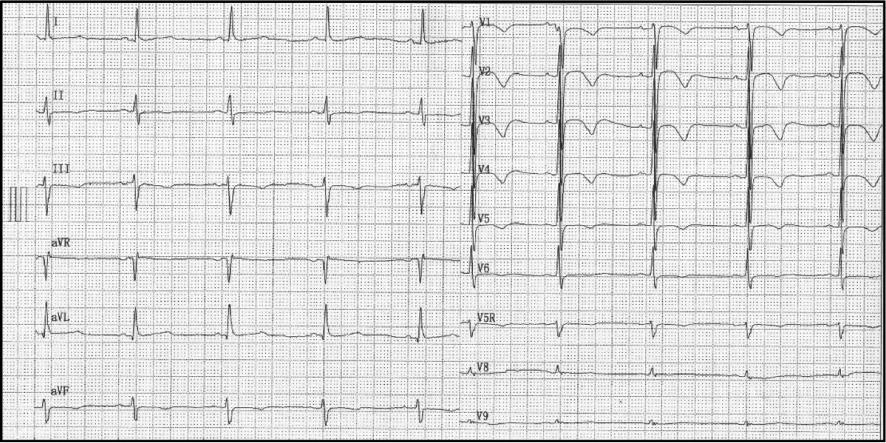
The first 12-lead ECG within 48 hours of his admission showed a heart rate of 84bpm, a normal PR interval of 140 ms, a QRS duration of 102 ms, and a prolonged QT/corrected TQ (QTc) interval of 448/526 ms. The T wave was biphasic especially in thoracic leads V2 to V4 (Figure 2). Twenty-four-hour Holter monitoring showed that the basic heartbeat was in sinus rhythm with fluctuations of the heartbeat in the normal range. A randomly occurring premature ventricular beat in the form of coupling, four short periods of ventricular tachycardia that lasted from 3 to 15 heartbeats for each period, and continuing ST-T changes were also recorded (Figure 3). In the 12-lead ECG recorded 6 days later, sinus tachycardia was detected. T-wave morphological changes such as low-amplitude or inverted T wave could be seen in most leads. The QT/QTc interval was 334/446 ms (Figure 4). However, another ECG recorded 1 week later still displayed an abnormal T wave with a QT/QTc interval of 530/568 ms (Figure 5). A bedside echocardiogram and enhanced magnetic resonance imaging, which did not show any cardiac structure abnormality, ruled out any cardiomyopathy disorders such as hypertrophic cardiomyopathy, dilated cardiomyopathy, or myocarditis (Supplementary Figure 1). Several periods of epilepsy, uncontrollable jerking movements of the arms and legs, and loss of consciousness or awareness occurred during his hospitalization, which may have contribution to his shortness of breath. CT demonstrated inflammation of his lungs, and no structure abnormality was found in the head. Respiratory system function gradually improved after transoral tracheal intubation and use of combined antibiotic drugs. Life support and antiepileptic and anti-inflammation treatment were used to relieve his suffering. Prolonged QT intervals in several ECGs led us to suspect LQTS. Implantable cardioverter-defibrillator implantation and immediate genetic testing, which are used to avoid further deleterious arrhythmia events and the diagnosis of LQTS, respectively, were refused by his parents after they had been fully informed of the potential risk. Propranolol at a dose of 2 mg/kg was used to control the constant fast heartbeat. After symptom relief, his parents asked to leave the hospital. (Before their departure, the boy was advised not to undertake rigorous exercise and his caretakers were taught cardiopulmonary resuscitation). Home management was done according to guidelines [6]. Propranolol at a dose of 1 mg/kg, twice a day, was suggested for him. Regular ECG or Holter monitoring was required each month after discharge. A 12-lead ECG 2 months after discharge showed a sinus heartbeat of 59 bpm and a QT/QTc interval of 456/454 ms with minor T-wave alternans (Figure 6).



Multiple Short Periods of Ventricular Tachycardia were Recorded by Holter Testing of the Proband.

Six Days After Holter Testing, Sinus Tachycardia was Recorded on a 12-Lead ECG of the Proband.

Clinical Features of Family Members
The proband’s father had a history of syncope without a clear memory of what caused it. His 12-lead ECG showed abnormal T wave morphology and a QT/QTc interval of 480/462 ms (Figure 7). The proband’s 50-year-old aunt also reported a history of syncope especially when she was young, with no inducing factors. Her 12-lead ECG revealed a sinus tachycardia with a normal QT/QTc interval of 440/405 ms. However, detailed clinical information on them is not available. The rest of the family members reported no symptoms.
Genetic Analysis
The c.1898A>C mutation in KCNH2 (Figure 8), an A-to-C nucleotide substitution at position 1898 resulting in the change of asparagine to threonine at position 633, and the c.916dupA mutation in JUP (Figure 9), a duplication of nucleotide A at position 916 leading to a frameshift effect after amino acid 366, were identified in the proband. The c.1898A>C mutation in KCNH2 was identified only in his paternal line relatives II-2, II-1, and II-5, whereas the c.916dupA mutation in JUP was detected in II-6, II-7, and III-4, all of whom are his maternal line relatives. Detailed clinical and genetic characteristics of family members are listed on Table 1.


Clinical and Genetic Characteristics of Family Members.
| Family member | Sex | Age (years) | KCNH2 c.1898A>C | JUP c.916dupA | HR (bpm) | QT/QTc interval (ms) | Phenotype |
|---|---|---|---|---|---|---|---|
| I-4 | Female | 69 | +/− | −/− | 60 | 424/424 | Syncope |
| II-1 | Female | 50 | −/− | +/− | 51 | 440/405 | Syncope |
| II-2 | Female | 46 | −/− | +/− | 67 | 376/392 | Asymptomatic |
| II-3 | Male | 46 | +/− | −/− | 54 | 480/462 | Syncope |
| II-5 | Male | 35 | −/− | −/− | 76 | 386/442 | Asymptomatic |
| II-6 | Male | 40 | +/− | −/− | 86 | 340/408 | Asymptomatic |
| III-1 | Female | 20 | −/− | +/− | 69 | 392/420 | Asymptomatic |
| III-2 | Male | 15 | +/− | +/− | 88 | 530/568 | Syncope, SCD |
| III-3 | Male | 8 | −/− | −/− | 81 | 352/409 | Asymptomatic |
| III-4 | Female | 12 | −/− | −/− | 69 | 400/429 | Asymptomatic |
| III-5 | Female | 10 | −/− | −/− | 87 | 380/458 | Asymptomatic |
HR, heart rate; QTc corrected QT; SCD, sudden cardiac death.
Discussion
We report a novel case of digenic mutation, KCNH2 c.1898A>C and JUP c.916dupA, in an LQTS-affected patient. The proband, a 15-year-old boy, had experienced syncope and seizure many times. The same symptoms, which lasted longer than previously, recurred after exhaustion during physical exercise. After his admission to the local hospital, cardiac arrest occurred three times and the ECG revealed torsades de pointes and ventricular tachycardia. He was fortunate to be rescued by cardiopulmonary resuscitation performed by physicians. Several of his 12-lead ECGs showed a prolonged QT/QTc interval. The longest QTc interval recorded was 526 ms, which is much longer than the diagnosis standard for LQTS. The low-amplitude notched and biphasic T wave, although not identified in every ECG, led to our suspecting type 2 LQTS (LQT2) [7].
High-throughput sequencing, which identified a c.1898A>C mutation in the KCNH2 gene, led to the diagnosis of LQT2 in the proband. Encoding a pore-forming α-subunit of the cardiac K+ channel, KCNH2, a human ether-a-go-go–related gene, also named HERG, has been proved to be an LQT2-related gene. The novel missense mutation c.1898A>C (p.N633T), alongside other reported mutations upstream and downstream of it, resulting in p.P632S, p.P632A, p.T634I, and p.E637K, is located in the hotspot variant region of the S5-loop-S6 region [8–10]. The S5-loop-S6 region is a transmembrane pore region of the potassium channel encoded by KCNH2. Moss et al. [11] showed that LQT2 patients who carry mutations on KCNH2 that involved in the pore region of the potassium channel are more likely to have malignant arrhythmia–related cardiac events at an earlier age than those who do not. In our study, the proband had experienced syncope since he was 10 years old. During his stay in the hospital, ventricular tachycardia, torsades de pointes, and cardiac arrest all occurred, which is in accordance with the study of Moss et al. His 69-year-old grandmother, carrying the c.1898A>C mutation in the KCNH2 gene, leads a normal life with no syncope or any history of cardiac arrhythmia. The proband’s father was also found to carry the same mutation. One of his 12-lead ECGs showed a notched T wave in the V1, V2, V3, and V4 leads. The morphology of T wave changes is helpful in the diagnosis of LQTS and in identifying patients at high risk of LQTS [12]. In the case of the proband’s father, further clinical assessment is needed to avoid any deleterious cardiac events.
Seizure and convulsion for minutes, the proband’s first symptoms rather than deleterious arrhythmias, can easily lead clinicians to the wrong diagnosis of epilepsy. In this case, the proband was initially treated as having epilepsy and was given antiepileptic drugs. However, little improvement was observed. Cardiac arrest occurred shortly after he had received the antiepileptic medication, but the reason for this is hard to explain. Experimental results obtained in human embryo kidney 293 cells by Danielsson et al. [13] indicated that some antiepileptic drugs such as phenytoin and phenobarbital, by blocking the rectifying potassium current (IKr) through IKr channels, which are encoded by the KCNH2 gene, can exert an arrhythmogenic effect, especially in some predisposed patients. Several studies have shown the seizure phenotype was more common in LQT2 than in other types of LQTS, especially caused by alterations in the pore region of the potassium channel resulted from KCNH2 mutations [14]. KCNH2 was originally isolated from a hippocampal cDNA library [15]. It is therefore plausible that seizures in LQT2 patients are in part a result of the effects of mutations in KCNH2, which could disturb the balance of potassium by having an influence on potassium channels in both the heart and neuronal tissues.
The JUP gene, encoding the protein junction plakoglobin, which plays a critical role in the formation of two important cell-binding and cell-supporting substances, adherens junctions and desmosomes, respectively, functions mainly in heart and skin. Mutations of the JUP gene are associated with arrhythmogenic right ventricular cardiomyopathy (ARVC), an inherited cardiac disorder with a devastating clinical course of sudden cardiac death in young people and athletes [16, 17]. The high-throughput genetic testing identified a c.916dupA mutation of JUP in the proband and his mother and aunt. This novel mutation, resulting in frameshift translation of a nucleotide after position 916 due to the additional duplication of A in exon 6 of the JUP gene, was not found in the 1000 genomes database, dbSNP, or the Exome Variant Server database (http://evs.gs.washington.edu/EVS/). As far as we know, this novel mutation of JUP is the first reported to coexist in LQT2 patients with a KCNH2 mutation. It is not clear whether this mutation had any contribution to the proband’s malignant arrhythmias and seizure as no major heart structure abnormality, only a sign that the smooth level of ventricular muscle is relatively low was observed from cardiac enhanced magnetic resonance imaging of the proband. His aunt, II-1, who carries the same c.916dupA mutation of the JUP gene and who experienced sudden syncope without any advanced warning many times when she was young, exhibited only sinus bradycardia on the 12-lead ECG. The proband’s mother, another JUP mutation carrier, who has no memory of heart uncomfortableness, had normal heart structure and function on two model echocardiograms and left anterior branch block on ECG.
Compound mutations, two or more mutations in the same or different LQTS-related genes, are believed to account for 5–11% of the genotyped LQTS population.
In our study, the proband’s first experience of syncope was when he was 10 years old. His devastating arrhythmias and sudden cardiac arrest may be connected to the two novel mutations in different genes because his family members who have only one of the two mutations have a trivial or even no phenotype.
The incomplete phenotype in the family members who carry the same KCNH2:c.1898A>C mutation suggested gene mutation may not be the only factor contributing to the expression of disease. Indeed, about 10–35% of LQTS mutation carriers displayed borderline or even no clinical signs of disease [18]. Besides gene mutation, many factors, such a gene modifiers, age, sex, and environmental differences, can also influence the disease phenotype [19, 20].
Multiple mutations account for a small fraction of all mutations in genetic cardiovascular diseases, and individuals with multiple mutations often have a severer phenotype than single-mutation carriers [21]. This led us to wonder whether the digenic heterozygosity may have contributed to the proband’s severe phenotype. In this family, the KCNH2 mutation carrier’s age and lifestyle, such as exercise intensity, and other unknown factors may also result in a diverse phenotype. Some studies claimed SNPs may play a role in the incomplete phenotype among carriers of the same mutation [22, 23], but this was not elucidated in our study. Practically it is hard to explain the incomplete phenotype caused by the same mutation by use of any of the possible factors alone. The combination of each factor results in the diverse phenotype.
LQTS patients who carry two or more mutations of the same gene can have a higher risk of life-threatening cardiac events than those with a single mutation or no mutation [21, 24, 25]. However, research on LQTS patients who carry an LQTS gene mutation plus another hereditary arrhythmia gene mutation is lacking. We are not sure whether two different inherited arrhythmia gene mutations could have overlying effects where mutation in the two different genes may result in the same symptom like syncope.
In this study, regular follow-up is needed for the proband and his family members. It would have been good for him to have received an implantable cardioverter-defibrillator to avoid lethal arrhythmia. Furthermore, reproduction advice was given with regard to the health of further generations of the family.
Conclusion
We described digenic heterozygous mutation, c.1898A>C in the LQTS-related gene KCNH2 and c.916dupA in the ARVC-associated gene JUP, in a Chinese LQTS patient. The genotype expanded the spectrum of LQT2 mutations and enriched our understanding of the relationship between phenotype and compound mutations.