
BACKGROUND
Insects are well known as vectors for pathogen transmission. Several studies on the bacterial diversity of ticks have been conducted to reveal the transmission potential of bacterial infections, given that ticks were the first arthropods to be identified as pathogen vectors and that, alongside mosquitoes, they are recognized as a major arthropod vector of diseases affecting both humans and domestic animals worldwide [1]. Many tick-borne diseases have been reported, including Lyme borreliosis caused by Borrelia burgdorferi, Mediterranean spotted fever caused by Rickettsia conorii, Siberian tick typhus caused by Rickettsia sibirica subsp. sibirica, Q fever caused by Coxiella burnetii, and tick-borne encephalitis caused by Flavivirus [1,2]. A single tick bite carries an alarming risk of inoculation with several pathogens, because ticks typically contain diverse pathogenic microbial communities [1]. The tick Dermacentor nuttalli is an ectoparasite highly specialized in hematophagy and a small arthropod of the family Ixodidae of the subclass Acarina [3]. D. nuttalli is widespread in China, Eastern Siberia, and Mongolia, with an average lifespan of 86 days at room temperature and 1 year or more in nature [4,5]. D. nuttalli are triple-host ticks that overwinter mainly as starving adults, which infest large mammals in the spring; nymphs and larvae emerge in summer and fall and parasitize various rodents [6]. These ticks can transmit protozoan pathogens, most commonly Babesia [7–9]. Additionally, several well-known tick-borne zoonosis, such as tularemia, tick-borne rickettsiosis, Lyme disease, and tick-borne encephalitis, have been reported [4,10,11]. The spotted fever group rickettsiae in particular have been detected in D. nuttalli at high percentages and are carried by both adults and nymphs [10,12,13]. D. nuttalli is considered an Anaplasma ovis and lymphocytic choriomeningitis virus vector [14,15].
This study aimed to evaluate the bacterial diversity of adult ticks that were not feeding on large mammals and to detect potential pathogens, particularly Brucella spp., because ticks can act as vectors for brucellosis [16,17]. According to the life cycle of D. nuttalli, starving adult ticks emerge from the soil and rodent burrows, where they survive through winter, then climb high up vegetation in spring and wait for hosts, such as cattle and sheep passing by. They then feed and absorb blood until becoming engorged, and lay eggs in the soil. Considering the above life cycle, we collected D. nuttalli ticks from vegetation in March and April of 2017 to investigate the bacterial microbiota that they contained. Pathogens carried by these ticks are likely to be transmitted to humans. Culturomics and 16S rDNA amplicon sequencing were applied to identify potential bacterial agents carried by the unfed ticks [3].
METHODS
Sampling
During the spring (March and April) of 2017, questing adult D. nuttalli were collected as they emerged from the soil and rodent burrows and moved to vegetation. The ticks were manually sampled from the wild in Guertu County, Wusu City, which is located in the sylvosteppe of the western Tianshan mountainous areas in Xinjiang Province, China. The area spans 3894.25 km2 and has an average elevation of 575 m; the mean annual temperature is ~6.7°C. We collected ~8,000 adult ticks that were not feeding on parasitizing hosts. All ticks were identified as D. nuttalli on the basis of morphological characteristics [18]. Samples were selected to investigate the ticks’ microbial diversity without contamination due to feeding on mammals. All ticks were randomly divided into 36 groups of 200–300 individuals each and were stored in 10 mL tubes at 4°C until further examination. During the preservation period, an adequate quantity of water was added to support the survival of the ticks.
Surface sterilization and DNA extraction
Before DNA extraction and cultivation were performed, each tube of ticks (200–300 individuals) was submerged in a 75% ethanol solution for 30 s to remove any potential microbiological contamination on the ticks’ surfaces and then thoroughly rinsed with sterile phosphate-buffered saline (PBS) three times before crushing [19,20]. The ticks were ground into powder, which was suspended in 5 mL of sterile PBS. The suspension was used to extract the entire genome of the microbiota contained within the ticks with a commercial DNA extraction kit (QIAamp DNA Mini kit®) as previously described [21]. After extraction, the quality of the DNA solutions was determined with agarose gel electrophoresis, and the purity and concentration were assessed with the photodensitometry method.
16S rDNA amplicon sequencing
Amplification of the V4–V5 hypervariable regions of bacterial 16S rDNA was performed with a conventional polymerase chain reaction (PCR) procedure [22]. To eliminate contamination, sterile double-distilled water was used as a negative control in each assay. The primers 515F (5′-GTG CCA GCM GCC GCG GTA A-3′) and 907R (5′-CCG TCA ATT CCT TTG AGT TT-3′) used in the amplifications also included an 8 bp multiplex barcode and Illumina adapters. Amplifications were performed on a Bio-Rad T100 gradient thermal cycler (Bio-Rad, CA, USA), with 1 min of pre-denaturation at 98°C, 30 cycles for 10 s of denaturation at 98°C, 30 s of annealing at 50°C, 30 s of extension at 72°C, and a final extension step of 5 min at 72°C. An equimolar mix of each amplicon was detected with electrophoresis on a 2% agarose gel, and the target band was then purified with a GeneJET gel extraction kit (Thermo Scientific, USA). The products were prepared for sequencing. The libraries were constructed with an Ion Plus Fragment Library Kit for 48 reactions (Thermo Fisher, USA), then quantified with a Qubit instrument and Q-PCR. Paired-end sequencing of the qualified libraries was performed with the Illumina HiSeq2500 PE250 platform.
In the analysis of the V4–V5 hypervariable regions, sample data were separated according to the barcode sequences. The reads for each sample were spliced with FLASH after truncation of the barcode and primer sequences, thus yielding the raw tag data. To produce high-quality tags, denoted clean tags, the raw tags required rigorous filtering, including truncation and length filtration. Finally, after removal of the chimeras in clean tags, the effective tags were obtained and subjected to Operational Taxonomic Unit (OTU) clustering. Because all pools were from the same batch of samples, all DNA samples were mixed together for further analysis.
OTU clustering analysis
A total of 2,394,598 effective tags from all samples were used for OTU analysis to investigate the species composition diversity in the samples. OTUs are division units obtained by clustering 16S rDNA sequences with high similarity, and tags with 97% shared sequence identity were designated as species-level OTUs [23]. The taxonomic assignments of OTUs were performed with Uparse v7.0.1001. Simultaneously, the sequence with the highest frequency in each OTU was selected as the representative sequence of the OTU according to the Uparse algorithm principle, after which species annotation analysis was conducted with the representative sequences selected above according to the Mothur method and SILVA’s SSUrDNA database (set threshold of 0.8–1). Consequently, OTU taxonomic information was obtained, and the sample community composition was counted at each classification level (referring to kingdom, phylum, class, order, family, genus, and species). Finally, the data were homogenized by the group with the fewest effective tags in all samples, and subsequent alpha diversity analysis (rarefaction curve, Chao1, and Shannon index) was performed on the data after homogenization (beta diversity analysis was not conducted because the same samples were used) [21].
Culturomics
Sample pretreatment in culturomics was conducted as described in the DNA extraction section. After sterilization with 75% ethanol and washing with sterile PBS, sampled ticks were ground into powder and suspended in 10 mL PBS. Brucella-specific immunomagnetic beads were used to capture bacteria of the genus Brucella, and Brucella-selective solid medium was used for culture. In contrast, 1/10, 1/100, and 1/1000 dilutions of ground tick solution were applied to spread plate cultures on Tryptone Soya Agar (OXOID, UK) and Brain Heart Infusion (Becton Dickinson, USA) solid media for the isolation of various bacteria. All culture media were incubated at two temperatures (26°C and 37°C) and under aerobic/anaerobic conditions for 3–7 days [24].
Bacterial identification with MALDI-TOF mass spectrometry and 16S rDNA sequencing
When colonies appeared, four colonies with the same morphology were selected randomly, and three-zone inoculation was performed for further isolation. Bacteria were identified with matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (BrukerDaltonics, Germany; AutoBio, China) according to the manufacturer’s recommendations [25,26]. Additionally, for isolates that could not be identified by MALDI-TOF, PCR was performed on 16S rDNA with the universal primer pairs 27F/1492R, and the products were analyzed with BLAST from NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch) and RDP (https://rdp.cme.msu.edu/classifier/classifier.jsp).
RESULTS
Bacterial microbiota in D. nuttalli with 16S rDNA amplicon sequencing
The quality control report indicated that the DNA quality of group P1 was too low to be applied to further sequencing, and the double-distilled water control was negative (not shown); therefore, only the other 35 groups of tick samples were used in 16S rDNA amplicon sequencing analysis. The overall number of clean data (that is, the number of effective tags after filtration that were used in OTU clustering and further analysis) was 2,394,598, of which 2,373,679 tags were clustered into 461 OTUs with species annotation information (Fig 1A). Nevertheless, 20,882 unique tags (frequency of 1, which could not be clustered into OTUs), and 37 unclassified tags (with no comment information available) were not subjected to subsequent analysis. Furthermore, a substantial fraction of effective tags could not be aggregated to the species level because of an identity higher than 97% among species from the same genus (Fig 1B). A rarefaction curve assessing alpha diversity was constructed to describe the diversity of samples within the group (Fig 2A). The curve not only directly reflected the adequacy of the amount of sequencing data but also visually indicated the richness and uniformity of species in the sample, as verified by the Chao1 and Shannon index (Fig 2B and C). According to alpha diversity, the results of OTUs clustering indicated that the amount of sequencing data were adequate, and the species richness was high; however, the homogeneity was poor because of the presence of several dominant species, such as Pseudomonas spp. together with many low-abundance species. The bacterial microbiota of D. nuttalli was dominated by two genera (Fig 3): Pseudomonas (phylum Proteobacteria, class Gammaproteobacteria) and Coxiella (phylum Proteobacteria, class Gammaproteobacteria), which represented as much as 71% of the total D. nuttalli microbiota (Table S1).

OTU clustering and classified statistics of effective tags.
(A) OTU clustering and tag annotation statistics. Total tags: the total number of effective tags in the sample (valid data for subsequent analysis); unclassified tags: tags with no annotation information available; taxon tags: tags with annotation information available that were used in OTU analysis; unique tags: tags with a frequency of one that could not be clustered into OTUs (and could not be used for subsequent analysis); OTUs: the number of OTUs obtained. (B) Classified statistics of effective tags. The value indicates the number of tags clustered to various classification levels, including kingdom, phylum, class, order, family, genus, and species.
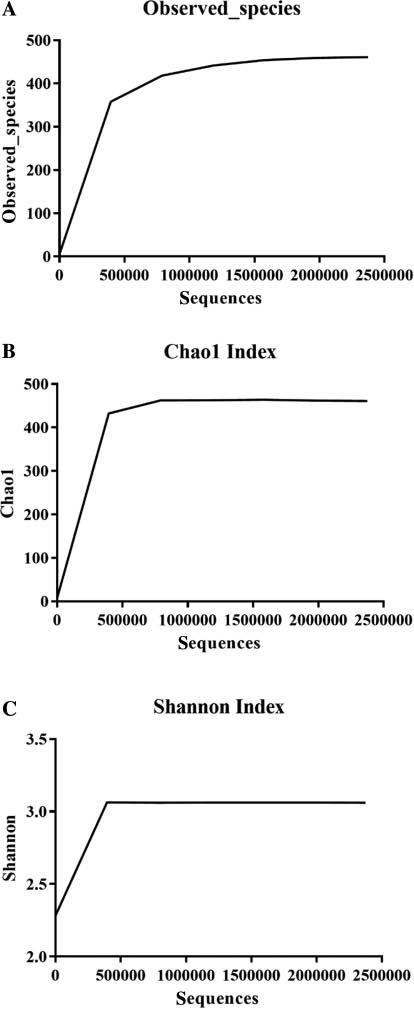
Alpha diversity indicating microbial community diversity within samples.
(A) The number of observed species. The horizontal axis represents the number of sampled sequences, and the vertical axis represents the number of observed species. (B) Chao1 index with the same horizontal axis and vertical axis representing the Chao1 index value. (C) Shannon index with the same horizontal axis and the vertical axis representing the Shannon index value.

Column chart of bacterial diversity, showing the relative abundance of the top ten genera.
The vertical axis represents the relative abundance of each of the top ten genera. “Others” (in yellow) represents the proportion of all other genera, excluding the top ten genera.
In species taxonomy analysis of the microbiota within tick samples, the top ten genera with maximum relative abundance, which were of interest by default, were selected for taxonomic tree building (Fig 4). Several species in the taxonomic tree had an abundance value of zero, such as Pseudomonas psychrotolerans, because the abundance of the species was too low to be displayed on this tree. Furthermore, the percentage inconsistency of a genus and its species was due to the presence of tags with no available species-level identification, as shown in Fig 1B.

Taxonomic tree constructed on the top ten genera within the sample.
Circles with different colors represent the seven taxonomic levels, corresponding to the left legend; the circle size represents the relative abundance of taxonomy; the two numbers below the taxonomic names indicate the relative abundance percentage: the first number indicates that the taxonomy accounts for all taxonomies of the sample, and the second number represents the percentage of taxonomy accounting for the selected taxonomies used for tree building.
Bacterial microbiota in D. nuttalli determined with culturomics
In total, 284 bacterial colonies were isolated and identified with MALDI-TOF mass spectrometry and 16S rDNA sequencing. MALDI-TOF mass spectrometry was used to identify 274 isolates from 43 bacterial species (Table 1); ten isolates failed to be identified. The results of 16S rDNA PCR product sequencing and BLAST analysis revealed that the remaining isolates belonged to four species: Microbacterium phyllosphaerae, Microbacterium testaceum, Janibacter hoylei, and Sanguibacter inulinus, the latter two of which are newly identified genera added in culturomics. In summary, 46 species from 23 genera were isolated and identified through culturomics approaches. Pseudomonas accounted for the highest proportion, up to 34.5% and was followed by Pantoea and Bacillus, which accounted for 17.3% and 10.2%, respectively (Fig 5). At the species level, 49 Pantoea agglomerans were identified and ranked second to the most abundant bacteria, Pseudomonas koreensis, in agreement with genus-level statistics. Additionally, several bacterial species were identified with only one isolate, including Arthrobacter citreus, Janibacter hoylei, Brevundimonas vesicularis, Pseudoxanthomonas kaohsiungensis, and Serratia marcescens, possibly because of the unsuitable culture conditions. In the case of isolation in Brucella-selective cultivation, we were unable to cultivate any Brucella spp.. The negative results were consistent with the 16S rDNA amplicon sequencing data, which yielded negative results for Brucella spp. even at the genus level.
Genera and species of bacteria identified by culturomics.
| Genus | Species | Number* |
|---|---|---|
| Acinetobacter | Acinetobacter pittii | 2 |
| Arthrobacter | Arthrobacter citreus | 1 |
| Arthrobacter gandavensis | 2 | |
| Bacillus | Bacillus cereus | 5 |
| Bacillus infantis | 1 | |
| Bacillus pumilus | 21 | |
| Bacillus simplex | 1 | |
| Bacillus thuringiensis | 1 | |
| Brevundimonas | Brevundimonas vesicularis | 1 |
| Candida | Candida lipolytica | 8 |
| Cellulosimicrobium | Cellulosimicrobium cellulans | 3 |
| Corynebacterium | Corynebacterium glutamicum | 8 |
| Corynebacterium mucifaciens | 1 | |
| Delftia | Delftia acidovorans | 10 |
| Ewingella | Ewingella americana | 2 |
| Exiguobacterium | Exiguobacterium aurantiacum | 8 |
| Janibacter | Janibacter hoylei | 1 |
| Microbacterium | Microbacterium liquefaciens | 9 |
| Microbacterium oxydans | 5 | |
| Microbacterium paraoxydans | 1 | |
| Microbacterium phyllosphaerae | 7 | |
| Microbacterium testaceum | 2 | |
| Micrococcus | Micrococcus luteus | 5 |
| Paenibacillus | Paenibacillus amylolyticus | 2 |
| Pantoea | Pantoea agglomerans | 49 |
| Pseudoclavibacter | Pseudoclavibacter helvolus | 2 |
| Pseudomonas | Pseudomonas chlororaphis | 2 |
| Pseudomonas extremorientalis | 4 | |
| Pseudomonas fluorescens | 6 | |
| Pseudomonas frederiksbergensis | 2 | |
| Pseudomonas grimontii | 1 | |
| Pseudomonas kilonensis | 1 | |
| Pseudomonas koreensis | 57 | |
| Pseudomonas libanensis | 6 | |
| Pseudomonas marginalis | 1 | |
| Pseudomonas orientalis | 1 | |
| Pseudomonas putida | 5 | |
| Pseudomonas synxantha | 12 | |
| Pseudoxanthomonas | Pseudoxanthomonas kaohsiungensis | 1 |
| Rhodococcus | Rhodococcus erythropolis | 11 |
| Sanguibacter | Sanguibacter inulinus | 4 |
| Serratia | Serratia marcescens | 1 |
| Staphylococcus | Staphylococcus epidermidis | 1 |
| Staphylococcus nepalensis | 2 | |
| Staphylococcus warneri | 3 | |
| Stenotrophomonas | Stenotrophomonas rhizophila | 5 |
*: Number indicates the quantity of isolates.

Comparison of bacterial microbiota in D. nuttalli through two methods
In this study, the results obtained with the two methods were largely consistent and complementary. Proteobacteria was most abundant at the phylum level, accounting for 90.7% and 59.5% of the 16S rDNA amplicon sequencing and culturomics datasets, respectively. Other microorganisms were mostly from the phyla Actinobacteria and Firmicutes. Moreover, Pantoea, Microbacterium, and Rhodococcus (17.3%, 8.5%, and 3.9%, respectively) which were frequently isolated with the culturomics method, were detected in the amplicon sequencing datasets in relatively high proportions (6.4%, 1.7%, and 0.9%, respectively), thus demonstrating that the results obtained with the two methods were largely consistent with each other (Table 2). Nevertheless, many additional specific identifications were accomplished with each method. Overall, 237 genera were identified, 214 of which were specifically detected with 16S rDNA amplicon sequencing methods, 4 of which were identified only by culturomics methods, and 19 of which were common to both methods (Fig 6A). For instance, we obtained a relatively high proportion of Bacillus spp. (10.2%) through culturomics, although the Bacillus abundance in the 16S rDNA amplicon sequencing results was less than 0.01%. Therefore, culturomics can amplify some bacteria with lower abundance. Furthermore, in our culturomics methods, we did not apply culture conditions for isolating Coxiella and Rickettsia, which accounted for 17.2% and 4.8% of the sequencing datasets, respectively. At the species level, each method yielded additional specific identifications. The two methods identified 159 species, 133 of which were specifically detected by amplicon sequencing, 40 of which were isolated by culturomics, and only 6 of which were common to both methods (Fig 6B). In conclusion, each metho provided specific information for identifying microorganisms in D. nuttalli; thus, integrating the two methods is crucial for thoroughly understanding the microbiota of D. nuttalli.
Comparison of the top ten bacterial genera identified by 16S rDNA amplicon sequencing and culturomics.
| Genus | Sequencing | Culturomics |
|---|---|---|
| Pseudomonas | 53.83* | 34.51 |
| Pantoea | 6.43 | 17.25 |
| Microbacterium | 1.67 | 8.45 |
| Rhodococcus | 0.90 | 3.87 |
| Coxiella | 17.22 | NC# |
| Rickettsia | 4.75 | NC |
| Thermus | 2.08 | NC |
| Acinetobacter | 6.04 | LP$ |
| Serratia | 1.08 | LP |
| Brachybacterium | 0.80 | NC |
| Bacillus | LP | 10.21 |
| Delftia | 0.32 | 3.52 |
| Corynebacterium | LP | 3.17 |
| Candida | - | 2.82 |
| Micrococcus | 0.01 | 1.76 |
| Stenotrophomonas | 0.55 | 1.76 |
*: Number indicates the percentage of identified bacterial genus among all identified genera, according to the corresponding method.
#: NC indicates no cultivation conditions used for isolating the corresponding bacterium.
$: LP indicates the bacterium isolated with a lower percentage.
“-” Indicates that no sequence was detected.

Comparison at the genus level (A) and species level (B) of bacterial diversity identified by 16S rDNA amplicon sequencing and culturomics methods.
(A) The dark yellow portion of the Wayne diagram indicates the number of genera identified by 16S rDNA amplicon sequencing. The blue portion indicates the number of genera identified by culturomics. The intersection indicates the genera common to both methods. (B) The blue portion of the Wayne diagram indicates the number of species identified by 16S rDNA amplicon sequencing. The green portion indicates the number of species identified by culturomics, and the intersection indicates the number of species common to both methods.
We focused on the absence of Brucella spp. in the results from both approaches to assess the probability of D. nuttalli ticks acting as the vector of Brucella spp. through transovarial transmission, similarly to Dermacentor marginatus. Pseudochrobactrum saccharolyticum, a member of the family Brucellaceae, was the only species identified by 16S rDNA amplicon sequencing, and no Brucella were identified, even at the genus level. Similarly, we were unable to cultivate any species of Brucella. These results indicate that further research is needed to determine whether D. nuttalli ticks are the vector of Brucella.
DISCUSSION
Our study objective was to investigate the composition of the microorganisms that are carried by, and have a symbiotic relationship with, D. nuttalli ticks; that could be transovarially transmitted and present during all life stages of ticks; and that do not appear to be pathogenic in ticks [27]. To do so, we captured starved adult D. nuttalli ticks when they emerged on vegetation in the spring. We selected adult ticks before they parasitized mammals, because the microbiota in the ticks at this stage were almost certainly symbiotic, after having persisted for at least an entire winter without feeding. The pathogens carried by these ticks are relatively likely to be transmitted to humans, because ticks in that stage are ready to infest large mammals, including humans, and then lay eggs after becoming engorged through blood sucking [6]. Furthermore, when ticks mature from larvae to adults, bloodmeal feeding usually decreases the diversity of internal tick microbiota; however, continuous exposure to the field environment may counter this loss [28,29]. Zhang et al., through the collection method described above, first identified and isolated a novel D. nuttalli tick-borne phlebovirus, named Guertu virus, which can infect animals and cause symptomatic or even lethal disease in humans [30]. The samples were washed with 75% ethanol to remove possible contamination with bacteria attached to the ticks’ surface before further investigation, thus ensuring that the microorganisms identified with culturomics were all from the ticks’ internal microbiota.
In our study, the genus Pseudomonas dominated the bacterial microbiota of D. nuttalli, a finding inconsistent with those from several previous studies reporting that Francisella, Bacillus, Rickettsia, or Coxiella was the most abundant genus identified in several other types of ticks, such as Dermacentor variabilis, Amblyomma tuberculatum, and Haemaphysalis [21,31–36]. Dead and decaying ticks may account for this discrepancy, owing to the humid environment, which is optimal for Pseudomonas growth [37]. The genus Francisella was not detected in the study, and the genera Bacillus and Rickettsia accounted for less than 0.001% and ~5% of the total, respectively, possibly because different types of ticks were collected from different locations. In previous studies, spotted fever group rickettsiae have been detected in D. nuttalli at a high proportion of 67.4%, and potential novel Rickettsia species have been found in D. nuttalli [12]. Furthermore, Rickettsia raoultii is probably the predominant Rickettsia in D. nuttalli. In addition, R. raoultii, and Rickettsia sibirica subsp. Sibirica, which causes Siberian tick typhus, have been found in nymphs, and both unfed and engorged adults of D. nuttalli [2,10,12,38]. The causative agent of Q fever, Coxiella burnetii, has also been detected in both unfed D. nuttalli ticks and D. nuttalli ticks collected from livestock [39–41]. Unfortunately, cell culture was not applied to culturomics; therefore, specific species information on Rickettsia and Coxiella was not obtained.
Given that past research has primarily focused on engorged ticks collected from animals, and that most studies have used either 16S rDNA amplicon sequencing or culturomics approaches, we first applied both methods in this study to thoroughly investigate the bacterial diversity of unfed adult D. nuttalli ticks. Recently, 16S rDNA amplicon sequencing has become a commonly used approach for microbial identification, because of its advantages in identifying low-abundance and non-culturable bacteria. This method has been used in investigations of the bacterial communities in ticks [19,21,33,34,42]. However, the method based on next-generation sequencing alone is insufficient for gaining comprehensive insight into the bacterial communities associated with D. nuttalli ticks, because of the inherent limitations in taxonomic resolution when bacteria contain ≥97% sequence similarity and significantly different cut-off values between species and genera [43,44]. Furthermore, 16S rDNA amplicon sequencing has generated countless sequences that have not been assigned to known microorganisms, thereby necessitating the successful culture of new isolates [45]. Thus, we applied culturomics methods in our study, because combining the results would aid in assessing microbial diversity in pathogen-carrying insect vectors [3,24,46,47]. Indeed, more comprehensive information on microbiota in D. nuttalli ticks was obtained. For instance, the sequencing method results revealed that Pseudomonas was the most abundant genus in the bacterial community of D. nuttalli ticks, accounting for more than 50%, whereas Pseudomonas spp. accounted for less than 0.1% at the species level, owing to the limitations in taxonomic resolution. Complementarily, culturomics revealed results consistent with those of 16S rDNA amplicon sequencing in terms of the most abundant genus. A total of 12 Pseudomonas sp. were cultured and identified, thus demonstrating that culturomics can markedly enhance the identification resolution at the species level. Furthermore, although amplicon sequencing detected 233 genera and 113 species in this study, culturomics specifically identified 4 genera and 40 species, thus implying that culturomics may provide information on unassigned tags or OTUs obtained from amplicon sequencing. Therefore, our study indicates that although culturomics lacks high sensitivity to fastidious bacteria, it can still be an effective supplement to 16S rDNA amplicon sequencing in the investigation of the microbiota in tick samples.
To our knowledge, several bacteria isolated in this study are pathogens capable of causing bacteremia, such as Brevundimonas vesicularis, Acinetobacter pittii, and Exiguobacterium aurantiacum; however, these pathogens were all discovered in nosocomial infections [48–63]. Other nosocomial infections associated with our isolates were ventriculoarterial shunt infection caused by Staphylococcus warneri [64], pneumonia caused by Acinetobacter pittii [65], and line-related sepsis caused by Delftia acidovorans [66]. Because these microorganisms can cause diseases, such as bacteremia in nosocomial infections, we believe that they could potentially infect humans through tick bites.
The identification of Brucella spp. was performed because no vector for Brucella has been discovered in nature, and ticks can function as a vector for the transmission of Brucella to animals [16]. Given that enrichment with Brucella-specific immunomagnetic beads, cultivation on Brucella-selective medium, and 16S rDNA sequencing all yielded negative results, we hypothesized that Brucella is carried by only several other tick species in nature, such as Dermacentor marginatus, which can transmit Brucella transovarially [17]. Because of these negative results, further investigation is required to confirm whether D. nuttalli ticks are the vector of Brucella.
The investigation of potential pathogens carried by natural vectors is important. Here, we applied 16S rDNA amplicon sequencing and culturomics methods to assess the diversity of bacterial microbiota in unfed adult D. nuttalli ticks. A total of 237 genera and 157 species were identified in the study. The results indicate that the combined application of the two methods to investigate microbiota is crucial and could provide more comprehensive information. Our study provides the first preliminary profile of the complete microbiota from unfed adult D. nuttalli ticks, thus providing a basis for further investigations regarding the roles of several pathogens, such as rickettsial endosymbionts in tick-borne diseases. Furthermore, our results indicate that isolates in culturomics can cause bacteremia and may suggest that new pathogens associated with ticks may remain to be discovered.
There are several limitations to this study. To potentially obtain more bacterial species, optimization of culturomics methods is needed, such as by increasing broth enrichment, extending the culture time, and growing bacteria in anaerobic conditions with more media. Furthermore, ethanol sterilization was used to kill bacteria on the ticks’ surfaces, but the remaining bacterial DNA might have yielded additional sequencing data beyond the data from tick bodies.