
The novel coronavirus SARS-CoV-2 has caused severe outbreaks worldwide since late 2019, thus leading to the COVID-19 pandemic and severely influencing health and economies globally. A variety of symptoms, such as pneumonia, can occur during infection. The virus was believed to have a strong ability to spread among people. The World Health Organization (WHO) declared the outbreak caused by SARS-CoV-2 as a global pandemic. As of November 22, 2021, more than 256,480,022 infections and 5,145,002 deaths had been reported worldwide [1].
A many mutations in the SARS-CoV-2 genome have been found, thus resulting in a variety of new variants and indicating that the virus continues to evolve. The mutations might affect viral fitness and transmissibility [2]. The study of viral variants relies on the sequencing of the viral genome. A total of 5,504,986 sequences have been sequenced and deposited in the Global Initiative on Sharing All Influenza Data (GISAID, https://www.gisaid.org/) database [3]. Various nomenclature systems established by research groups such as GISAID, Nextstrain (https://nextstrain.org/), and Phylogenetic Assignment of Named Global Outbreak Lineage (PANGO lineage) have been used to distinguish SARS-CoV-2 variants. A nomenclature system was proposed by the WHO for the major variants—Variant of Concern (VOC), Variant of Interest (VOI), and Variant Under Monitoring (VUM)—according to the Greek alphabet. In total, four VOCs (Alpha, Beta, Gamma, and Delta), two VOIs (Lambda and Mu), and seven VUMs had been designated as of November 2021. Among these variants, the Delta variant was the most dominant worldwide in the last half of 2021.
On November 26, 2021, the WHO announced that a novel variant (previously named B.1.1.529 by PANGO) has been designated as a VOC and named with the Greek letter Omicron (O). The variant is believed to be associated with the recent abnormal increase in new COVID-19 cases in South Africa and nearby countries. The mutant strain has been detected in many countries and regions outside southern Africa, including the United Kingdom, Canada, and France. A total of 125 sequences of the Omicron variant collected from South Africa, Botswana, Israel, Australia, Italy, Belgium, and the Hong Kong SAR in China have been deposited in the GISAID database (accession date: Nov. 28, 2021), and the earliest sequence was sampled on November 11, 2021. Many countries and regions such as the United States, the United Kingdom, Switzerland, and Singapore have issued travel restrictions for South Africa and surrounding countries.
To understand the viral sequence characteristics, the released viral genomes of the Omicron variant have been extensively analyzed and compared with the previous variants to predict its potential transmission ability and risk.
The sequences of the Omicron variant are structurally consistent with the reference strain of SARS-CoV-2 (Wuhan-Hu-1, GenBank accession number NC_045512.2). Compared with the reference sequence, the consensus sequence of the variant contains 44 amino acid substitutions, 6 amino acid deletions, and 1 amino acid insertion. These alterations comprise 29 amino acid substitutions, 3 amino acid deletions, and 1 amino acid insertion on the spike (S) protein (Fig 1). The S protein of the Omicron variant has approximately two or more times the number of mutations found in other variants, and these mutations are the most notable feature of this variant (Table 1). The phylogenetic tree constructed with the Nextstrain website [4] shows that the variant has relatively high similarity to the Alpha variant and is clearly located in a different branch from the currently dominant Delta variant (Fig 2).

The amino acid mutation sites on the S proteins of four variants of SARS-CoV-2.
The mutation sites on the S protein of SARS-CoV-2 are marked with red lines at the indicated positions. NTD: N-terminal domain; RBD: receptor binding domain; FP: fusion peptide; HR2: heptapeptide repeat sequence 2; TM: transmembrane domain.
Comparison of five variants of concern
| Variant | PANGO lineage | Number of conserved amino acid mutations | ||
|---|---|---|---|---|
| Total | S protein | RBD | ||
| Alpha | B.1.1.7 | 21 | 9 | 1 |
| Beta | B.1.351 | 16 | 8 | 3 |
| Gamma | P.1 | 22 | 12 | 3 |
| Delta | B.1.617.2 | 20 | 8 | 2 |
| Omicron | B.1.1.529 | 51 | 33 | 15 |
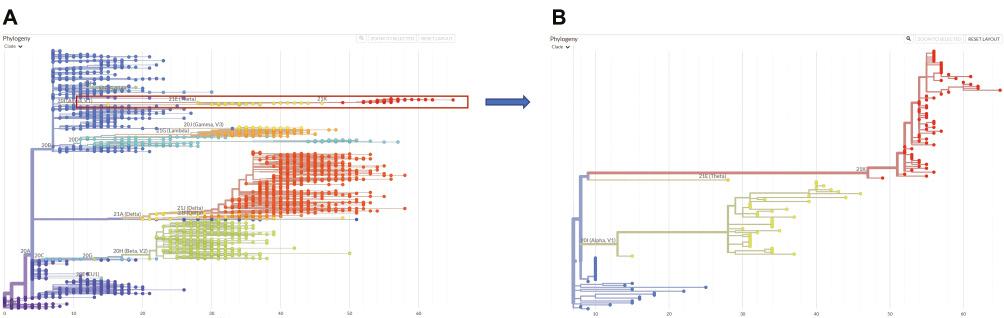
Phylogenetic reconstruction results of the Omicron variant.
A) Reconstruction results using representative sequences organized by Neherlab (https://neherlab.org/) and Nextstrain (https://nextstrain.org/). The sequences in the Omicron variant are shown as red dots. B) Zoomed-in results of the Omicron-variant-related sequences. Yellow dots indicate the Alpha variant, and blue dots indicate non-VOC and non-VOI sequences.
The S protein of SARS-CoV-2 contains 1273 amino acid residues, divided into two parts, S1 and S2, containing an N-terminal domain (NTD, 14–305), receptor binding domain (RBD, 319–541), fusion peptide (788–806), heptapeptide repeat sequence 1 (912–984), heptapeptide repeat sequence 2 (1163–1213), transmembrane domain (1213–1237), and cytoplasmic domain (1237–1273) [2]. The NTD region has been used as the target of partially neutralizing antibodies. The RBD region is associated with the binding of the virus to the host cell receptor human angiotensin-converting enzyme 2 (hACE2) and is the target region of most SARS-CoV-2 vaccines [5].
In the RBD of SARS-CoV-2, nine key residues (417, 449, 487, 489, 493, 500, 501, 502, and 505) are considered to be directly involved in the binding of S protein and hACE2 [6]. A total of 15 mutations have been found in the RBD of the Omicron variant, and four mutations (K417N, Q493R, N501Y, and Y505H) affecting these nine key residues might greatly enhance the binding ability of the virus to hACE2 and increase its infectivity. Among the mutations in the RBD, the N501Y mutation was previously identified in the Beta and Gamma variants, and is a widespread mutation. The binding affinity to the hACE2 receptor is nine-fold higher for S protein with the N501Y mutation than for wild type S protein [7].
For other mutations in the RBD region of the Omicron variant, the S477N mutation has been observed in the B.1.620 variant and has been associated with increased viral affinity toward the hACE2 receptor and with immune escape, together with the N439K and N501Y mutations [8–10]. The T478K mutation found in the Delta variant is associated with greater infectivity and less antibody neutralizing activity [11]. The Glu residue at position 484 has been reported to disrupt the hydrogen-bonding and salt bridge interactions associated with binding to neutralizing antibodies [12,13]. Moreover, mutation at this site might decrease the activity of neutralizing antibodies [14]. The E484K mutation has been detected in the Beta and Gamma variants, and the former VOIs Eta and Iota. The E484Q mutation has been found in the Alpha variant and the former VOI Kappa. Residue 484 in the Omicron variant bears an E484A substitution. The K417N mutation does not interact with hACE2 directly, but it facilitates more efficient binding to hACE2 [15,16]. The mutation has also been detected in the Beta and Gamma variants.
The D614G mutation in the S1-spike has been detected in all VOCs and VOIs, and is associated with greater binding affinity toward the hACE2 receptor and higher transmission ability [17,18]. The T95I mutation in the N terminal domain of S protein has been reported in the Iota variant and found to decrease neutralizing activity [19].
The 69/70 deletion is associated with immune escape and often causes diagnostic failure. The Y144 deletion alters immunogenicity [20]. The 142–144 deletion in the Omicron variant expands the Y144 deletion and might further decrease antibody neutralization reactions. The insertion of EPE at residue 214 has not been observed in other major variants of SARS-CoV-2. The NTD structural change due to the insertion has also been speculated to decrease neutralizing antibody binding ability.
The cleavage of S1 and S2 enables the fusion of S protein with the host. The S protein of SARS-CoV-2 can be primed by furin at the plasma membrane, owing to the insertion of PRRA amino acids in the cleavage site [21]. This cleavage site is associated with infectivity and pathogenicity. A total of three mutations (H655Y, N679K, and P681H) are present near the S1/S2 furin cleavage site. The N679K mutation was first reported in a VOC of SARS-CoV-2. The H655Y mutation, found in PANGO lineage P.2, plays an important role in conferring resistance to monoclonal antibodies and possibly may regulate S protein fusion and host cell entry in mammals [22]. The P681H mutation has also been reported in the variants Alpha and P.3. The mutation is a substitution of proline (P) to histidine (H) on the S-protein PRRAR furin cleavage site. The binding affinity of furin to S protein is enhanced by a conformational change and may even favor viral entry into host cells, thus increasing infectivity [23].
Beyond the S protein, mutation sites also exist in other proteins of the Omicron variant. The R203K and G204R mutations on the nucleocapsid protein are associated with enhanced expression of viral RNA and an increased viral load [24,25].
A variety of mutations are carried by the Omicron variant, which notably contains mutations at 33 sites and regions in the S protein that may enhance the spread of the virus, decrease neutralizing antibody binding ability, and increase viral RNA expression. The combined effects might confer strong advantages in the competition with other variants. Therefore, the Omicron variant has the potential to trigger a new global epidemic peak. More importantly, owing to the large number of mutation sites in the RBD region of the variant, there is a considerable probability that current vaccines may not have protective effects against this variant. People who have recovered from the COVID-19 may also infected again, thus causing new outbreaks in countries where the virus has already spread severely and widely.
According to epidemiological data, South Africa had experienced three large-scale outbreak peaks (Fig 3) [26]. The outbreak peaks were each caused by early strains of SARS-CoV-2, the Beta variant, or the Delta variant. The rapid increase in confirmed cases of COVID-19 in South Africa has indicated the formation of a fourth peak, and the number of infections has increased at a faster rate than that in the previous three peaks. These findings, combined with the genomic characteristics of the Omicron variant, indicate that this outbreak is highly likely to be caused by the wide spread of the Omicron variant.

Relationship between case number and variants in South Africa.
The daily case number in South Africa is displayed with a blue line, and the daily death number is displayed with a black line. The data and gallery were obtained from the WHO website (https://worldhealthorg.shinyapps.io/covid/).
Assessing the effects of the Omicron variant on the COVID-19 pandemic remains difficult. The increased transmission ability and replication potential of this variant may lead to wider spread, further enhancement of host adaptability, and possible recombination with other mutant strains to form super mutant strains. However, a trade-off between transmission ability and virulence might occur in this variant, given that the syndrome caused by infection with this variant has recently been reported to be relatively milder [27]. This mitigation might be due to a weaker host response caused by this variant’s large number of mutations; this possibility will require further validation through both clinical observations and experimental surveys.
Currently, urgent needs exist for rapid in-field detection methods for the Omicron variant and better understanding of its host interaction characteristics, to enable a prompt response strategy for this novel variant. Furthermore, investigating exactly where and how the Omicron variant originated will also be critical to aid in the prevention and control of not only the Omicron variant but also other variants generated through similar mechanisms in the future.
In summary, both the genomic and epidemiological characteristics of the Omicron variant suggest its potential for large-scale spread. The protective effects of the existing vaccine and the neutralization effects of existing antibodies on the variant must importantly be determined to gain the knowledge necessary to accurately evaluate the public health risk. Many countries and regions have introduced relevant prevention and control responses for the Omicron variant. Nevertheless, strategies for controlling Omicron transmission should be carried out by different countries acting together, to restrain the spread of this variant in a relatively early stage of appearance by building a network of intensive global cooperation.