
INTRODUCTION
Zoonoses (infectious diseases transmitted between animals and humans) are widely prevalent worldwide, and viral infections are the most urgent emerging zoonoses [1–3]. Greater 200 zoonoses that have been identified [4] include most of the emerging and re-emerging infectious diseases [5]. Based on pathogenic characteristics, zoonoses can be classified as bacterial, viral, fungal, parasitic, rickettsial, and mycoplasma zoonoses [6]. For example, plague [7], colibacillosis, and brucellosis [8] are zoonoses caused by bacteria and coronavirus disease (COVID-19), which is caused by severe acute respiratory syndrome (SARS) coronavirus 2 (SARS-CoV-2) [9], acquired immunodeficiency syndrome [10], Lassa fever [11], Ebola hemorrhagic fever [12], and Zika virus disease [13] are viral zoonoses, whereas cryptosporidiosis [14] and giardiasis [15] are zoonoses that arise from parasites. These diseases severely threaten human and animal health and can cause devastating global economic damage. Under the dual pressures of emerging and re-emerging zoonoses, many uncertainties remain in the prevention and control of these infectious diseases. Furthermore, infections due to these pathogens do not respond well to treatment and are associated with low survival rates; thus, early disease diagnosis and pathogen identification are particularly important in controlling the spread of zoonoses [3,16,17], with the current global SARS-CoV-2 pandemic a strong case in point.
The last two decades have witnessed a fast expansion in the repertoire of tools developed for pathogen detection. The clinical diagnosis of pathogens is generally based on the detection of pathogen-specific genes and proteins using techniques, such as reverse transcription (RT-)-polymerase chain reaction (PCR), RT-quantitative PCR (qPCR), enzyme-linked immunosorbent assay (ELISA), next-generation sequencing (NGS), fluorescence in situ hybridization (FISH), and chemiluminescence. These tools are very effective and have facilitated the evolution of diagnostics; however, traditional platforms, including serologic methods, which are only workable after antibodies have been produced [18,19], are time-consuming and can identify non-contagious cases as contagious (false-positive results), thus limiting the application of these methods in point-of-care testing (POCT). Pathogen detection tools based on antigen-antibody binding include ELISA [20], which is high in cost due to the heavy use of antibodies. Nucleic acid-based diagnostic methods, such as RT-PCR and RT-qPCR [21], are not convenient and require specialized equipment and trained personnel, although RT-qPCR is the gold-standard detection method for some RNA viruses, such as SARS-CoV-2 [21,22]. Nevertheless, nucleic acid-based tools have several advantages, including lower cost and higher sensitivity, over serologic methods [23,24]. Overall, these approaches are time-consuming, require strict operating procedures [25], are difficult in the rapid identification of pathogens, and cannot facilitate the interruption of pathogen transmission in real time, which is a challenge for global public health. In the setting of an increasing number of emerging and re-emerging zoonotic pathogens circulating among human populations, such as SARS-CoV-2, which remains as a pandemic since its emergence in late 2019, there is an urgent need for the development of new diagnostic tools that fulfill the affordable, sensitive, specific, user-friendly, rapid and robust, equipment-free, and deliverable to end-users needs [21].
Following the rapid evolution in our understanding of the clustered regularly interspaced short palindromic repeat (CRISPR-)-Cas systems, different Cas effector proteins have been used for the identification of zoonotic pathogens based on their unique characteristics. This novel platform is highly dependent on nucleic acids isolated from pathogens and detection based on nucleic acid amplification. Isothermal amplification methods, such as recombinase polymerase amplification (RPA), loop-mediated isothermal amplification (LAMP), and RT-LAMP, are effective for pathogen detection in low-resource settings [19,21,26–28]. The CRISPR-Cas-based diagnostic platforms utilize isothermal amplification techniques to detect pathogens for high-speed POCT with improved sensitivity.
Aptamers, which are short single-stranded artificial oligo (deoxy) nucleotides, have several advantages over antibodies, including smaller molecular weight and easier synthesis and screening [29]. Many aptamer-based biosensor detection platforms that are combined with physical, chemical, and electrochemical approaches are widely used for POCT. These pathogen detection platforms often involve multiple disciplines. Although CRISPR-Cas- and aptamer-based detection methods provide several advantages over conventional tools for pathogen detection, shortcomings remain. For example, CRISPR-Cas-based methods require nucleic acid extraction and amplification, whereas aptamer-based detection methods require aptamer screening, which is time-consuming (Table 1). Recent studies have introduced the combination of CRISPR-Cas- and aptamer-based platforms as a means to overcome these shortcomings (Table 1). These novel platforms exhibit robust detection capabilities and advantages over traditional methods and have potential utility for pathogen diagnosis and food health surveillance.
Advantages and disadvantages of tools for pathogen detection.
| Tools | Advantages | Disadvantages |
|---|---|---|
| Traditional | Accurate, sensitive, gold standard for particular pathogens | High cost, time-consuming, need for sophisticated equipment and skilled operators |
| CRISPR-Cas-based systems | Rapid, convenient, accurate, sensitive, robust, specific, time savings, reduce the false-positive rate, does not require sophisticated equipment, easy to operate, can be combined with other platforms, such as microfluidics and gene chip technologies | Need for cumbersome procedures (nucleic acid extraction and amplification), need a target site near the PAM sequence (Cas12), only recognize the RNA target (Cas13) |
| Aptamer-based systems | Convenient read out, accurate, low cost, high-binding specificity, and affinity, stable, wide target range, multimode readout | Screening process is difficult and requires significant time, not sensitive to clinical samples, leading to increased cost of screening |
| Combined CRISPR-Cas- and aptamer-based systems | Merits of both systems, aptamer-mediated enhanced activation of the Cas protein | Mainly focuses on the detection of pathogenic bacteria |
PAM, protospacer adjacent motif.
RAPID DETECTION PLATFORMS BASED ON CRISPR-CAS SYSTEMS
Discovery and evolution of the CRISPR-Cas systems
CRISPRs are short palindromic DNA sequences that were discovered in the Escherichia coli genome in 1987 [30]; CRISPRs are absent in eukaryotes and viruses [31]. The two components of the CRISPR-Cas systems are as follows: Cas proteins, which are CRISPR-associated endonucleases that cleave specific sequences; and small guide RNAs (sgRNAs), which are composed of CRISPR RNA (crRNA) and trans-activating RNA, recognize substrate DNA or RNA, and direct the Cas protein to the target site for editing. In nature, the CRISPR-Cas systems are part of the adaptive immune system of bacteria and archaea. Through the formation of RNA-guided endonuclease, CRISPR-Cas cleaves nucleic acids (DNA or RNA) that have invaded the prokaryote, thereby acting as a defense against foreign pathogens and plasmids [32–35]. The CRISPR-Cas systems involve three main phases of immunity: adaptation; expression/maturation; and interference [36–39]. As a result, specific Cas proteins record nucleic acid information in the CRISPR array following the invasion of foreign mobile genetic elements. The effector complex formed by the endonuclease recognizes the exogenous nucleic acid information previously stored in the CRISPR array and cleaves the invading extraneous elements of secondary infection.
The CRISPR-Cas systems can be categorized as class 1 and 2 based on genetic loci and the conserved sequences of the Cas protein. Class 1 CRISPR-Cas systems include types I, III, and IV, whereas class 2 CRISPR-Cas systems include types II, V, and VI (Table 2) [40,41]. Additionally, in contrast to class 1 CRISPR-Cas systems, which are composed of multiple proteins, class 2 CRISPR-Cas systems, which are composed of a single protein, hold promise for utility in a wide range of applications in genome editing, gene regulation, molecular diagnostics, in vitro pathogen detection, and DNA imaging. Class 2 CRISPR-Cas systems, especially those including Cas9 and Cas12a (former known as Cpf1), have been more extensively studied than class 1 CRISPR-Cas systems. The first Cas protein utilized in genetic manipulation was Cas9, which was purified from Streptococcus pyogenes (spCas9) [42]; however, activated Cas9 proteins, even those Cas9 proteins isolated from other bacteria, cannot be used for in vitro pathogen detection due to the lack of ability to non-specifically cleave nucleic acids. The subsequently identified Cas12a protein, however, has been recognized to perform this function. Interestingly, deactivated Cas9 (dCas9) is primarily used for pathogen detection based on its sequence-specific binding feature in the absence of cleavage activity [43,44]. Cas9 and Cas12a are the most extensively studied Cas proteins for gene editing and show promise for clinical application [45]. With the advent of Cas12, Cas13, and Cas14, several rapid detection techniques based on CRISPR-Cas systems have been explored. Notably, both Cas12 and Cas14 belong to the type V CRISPR-Cas system; the latter occupies only one-half of the amino acids of the former and does not require the identification of the protospacer adjacent motif (PAM) sequence, which is necessary for recognition by Cas9 and Cas12 [46]. Therefore, Cas14 may have more robust features and applications than Cas12.
Characteristics of class 2 Cas proteins.
| Cas protein | Domain | Protein length | Target molecular | PAM | Type of RNA | Trans-Cleavage |
|---|---|---|---|---|---|---|
| Cas9 | RuvC HNH | 1368aa | dsDNA | NGG | gRNA (crRNA+tracrRNA) | No |
| Cas12 | RuvC | 1300aa | dsDNA ssDNA | TTTV | crRNA | Yes (ssDNA) |
| Cas13 | Two HEPN | 1250aa | ssRNA | * | crRNA | Yes (ssRNA) |
| Cas14 | RuvC | 530aa | ssDNA | — | gRNA (crRNA+tracrRNA) | Yes (ssDNA) |
*Non-G nucleotide at the 3’ protospacer flanking site (PFS).
Gene editing and detection based on the type II CRISPR-Cas system
CRISPR-Cas9 can cleave double-stranded DNA (dsDNA) [32], which can be inserted into exogenous gene fragments for homologous recombination or non-homologous end-joining repair for gene knock-in or -out purposes [37,44,47], and can be used to successfully edit diverse genes by changing the sequence of gRNA to arbitrarily switch genes of interest. In addition, silence mutations in Cas9 that lead to dCas9 can be applied to diagnostics.
CRISPR-dCas9
The primary function of CRISPR-Cas9 is to splice target genes. Additionally, CRISPR-Cas9 can be applied to pathogen detection by exploiting its sequence-specific binding property that does not depend on cleavage activity (Fig 1). This detection procedure requires the introduction of two mutations to Cas9 (D10A and H840A mutations in the RuvC-I and HNH nuclease domains, respectively) [44,47]. The dCas9 harboring these silence mutations results in the removal of dsDNA cleavage activity but exhibits strong affinity for the substrate dsDNA [48] and can therefore regulate the expression of endogenous genes without gene editing. This feature of dCas9 has been used for labeling specific sequences. The CRISPR-dCas9 system can be developed for genetic screening, overexpression of genes of interest, regulation of target gene expression using transcriptional activators and repressors, and gene detection [49]. The majority of CRISPR-dCas9-based diagnostic tools can detect different target pathogens by changing the sequence of sgRNA.
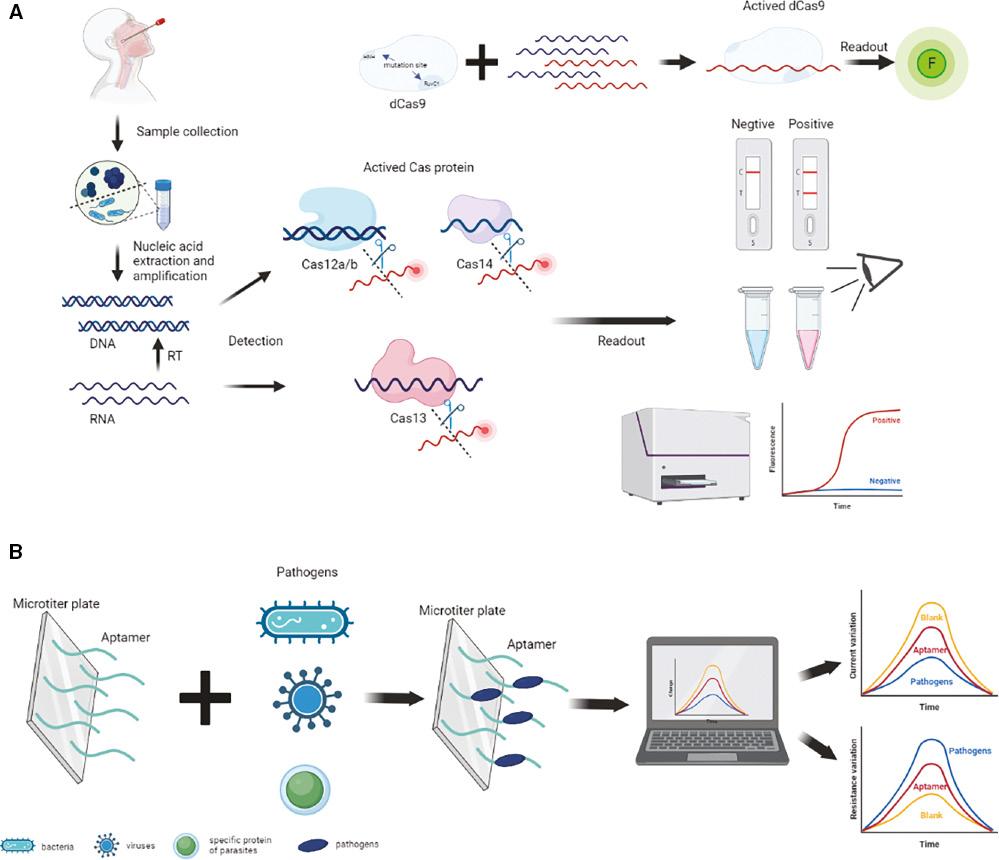
Schematic illustration of the diagnostic platforms. (A) Tools based on the CRISPR-Cas system for pathogen detection using collateral cleavage activity and sequence-specific binding of Cas12, Cas13, Cas14 and dCas9. The procedure involves sample collection, nucleic acid extraction and amplification, Cas protein activation, fluorescence probe degradation, and readout of the results. (B) Aptamer-based diagnostic tools for bacteria, viruses, and parasite-specific proteins. Aptamers capture the pathogen, causing a change in the current or resistance that indicates the presence or absence of the target pathogen. CRISPR, clustered regularly interspaced short palindromic repeat.
Assays based on CRISPR-dCas9 systems can potentially complement and enrich existing platforms used for pathogen detection. For example, the CRISPR-Tag system can detect herpes simplex virus 1 DNA in host cells [50,51] and CRISPR-mediated DNA-FISH has been used as a convenient, high-speed detection approach for methicillin-resistant Staphylococcus aureus [52]; however, CRISPR-dCas9-based detection tools have not become mainstream diagnostic platforms.
Establishing rapid detection platforms based on CRISPR-Cas trans-cleavage activity
Phylogenetic tree analysis has revealed that Cas12 and Cas14 belong to class 2 (type V) [53]. Unlike Cas9, Cas12a and Cas14 contain only one nuclease domain (RuvC [RuvC-I, RuvC-II, and RuvC-III]), which cleaves DNA, whereas Cas13 contains two HNH domains, which only cleave RNA [53–55]. Additionally, multiple Cas12 variants, such as Cas12b, Cas12e, Cas12f, and Cas12j, have been identified. Similar to Cas12a, Cas12b has been harnessed for the development of diagnostic tools [21]. Cas12e (formerly known as CasX) and Cas12j, have been reported to edit the human genome [56–59]. A recent study demonstrated a novel method using gene disruption and therapy using CRISPR-Cas12f, which was effectively packaged into an adeno-associated virus [45]. Given that dCas9, which has evolved from Cas9, can be used in diagnostics, whether these Cas12 variants can be applied to pathogen detection requires further investigation.
Notably, Cas12a, Cas12b, Cas13, and Cas14 possess collateral cleavage activity. The CRISPR-Cas systems can be activated to non-sepcifically cleave surrounding single-stranded DNA (ssDNA) or RNA following the recognition of predetermined specific sequences [53,55,60]. This collateral trans-cleavage activity is the basis for a highly sensitive and specific in vitro rapid detection platform that utilizes a single-stranded fluorescent reporter probe. The CasN-sgRNA complex, wherein CasN represents Cas12, Cas13, or Cas14, is combined with a target sequence that can activate the trans-cleavage activity of the Cas protein and cut a fluorescent probe, causing positive signals. Efficient degradation of the fluorescent probe can be used to confirm the presence of pathogens (Fig 1). CRISPR-Cas-based diagnostic tools convert the detection of target microorganism from a molecular level to a fluorescent signal that can be directly assessed by the naked eye or quickly identified by equipment, which is a more convenient, intuitive, and time-saving approach than conventional platforms.
In summary, the CRISPR-Cas systems have already been employed for the detection of disease-causing viruses, such as SARS-CoV-2 (Table 3), human papillomavirus (HPV) types 16 and 18, human immunodeficiency virus (HIV), Zika virus (ZIKV), and Dengue virus (DENV), and for the testing of food for organisms, such as E. coli, S. aureus, and Salmonella enterica serotype Enteritidis (S. enteritidis). Furthermore, the CRISPR-Cas systems have been harnessed to detect single nucleotide polymorphisms (SNPs), genetic mutations, and cancer (Table 4).
CRISPR-based diagnostic tools for rapid and accurate detection of SARS-CoV-2.
| Nuclease | Platform | Amplification method | Year | Refs. |
|---|---|---|---|---|
| Cas12a | DETECTR | RT-LAMP | 2020 | [28,61] |
| CRISPR-Cas12a-NER | RT-RAA | 2020 | [62] | |
| AIOD-CRISPR | RPA | 2020 | [63] | |
| iSCAN | RT-LAMP | 2020 | [21] | |
| STOP | RT-LAMP | 2020 | [64] | |
| OR-DETECRT | RT-RPA | 2021 | [65] | |
| M-CDC | RT-RPA | 2021 | [66] | |
| sPAMC | RPA | 2022 | [67] | |
| Cas12b | CASdetec | RT-RAA | 2020 | [68] |
| iSCAN (one-pot) | RT-LAMP | 2020 | [21] | |
| Cas13a | SHERLOCK | RPA/RT-RPA | 2020 | [69] |
| CREST | Amplification-free | 2021 | [70] | |
| Cas13b | SHERLOCKv2 | RPA | 2020 | [64] |
Reported CRISPR-Cas-based diagnostic tools.
| Nuclease | Platform | Model | Assessment | Testing time | Refs. |
|---|---|---|---|---|---|
| Cas12 | DETECTR | HPV16/18 | Fluorescence | Within 1 hour | [60] |
| Electrochemical DNA biosensor | Change of current (ΔI) | ∼1 hour | [71] | ||
| — | ASFV | Fluorescence | Within 2 hours | [72] | |
| Cas-gold | Gold nanoparticle-antibody strips | 1- or 2- minutes | [73] | ||
| HOLMES | JEV | Fluorescence | ∼1 hour | [74] | |
| HOLMESv2 | JEV, SNP | Fluorescence | ∼1 hour | [75] | |
| CRISPR-actuate hydrogels | Ebola | μPAD | ∼1 hour | [76] | |
| Hydrazone chemistry-mediated | P. aeruginosa | Fluorescence | — | [77] | |
| BM-TOA | EGFR mutations | Fluorescence | — | [78] | |
| Cas13 | SHERLOCK | E. coli, KPC, P. aeruginosa, ZIKV, DENV | Fluorescence | Within 2 hours | [79,80] |
| CRMEN | MERS, SARS, HIV, ZIKV, DENV, HCV, HPV4, influenza | Fluorescence | — | [81] | |
| SHERLOCK+HUDSON | ZIKV, DENV, YFV, WNV | Fluorescence | Within 2 hours | [82] | |
| — | Treponema pallidum | Fluorescence | 1-4 hours | [83] | |
| Cas14 | DETECTR-Cas14 | HBoV1 | — | — | [46] |
| HUDSON+Cas14 | SNP (iris color variability) | — | — | [46] |
Diagnostic tools based on the type V CRISPR-Cas system
DNA endonuclease-targeted CRISPR trans reporter (DETECTR), which is based on Cas12a coupled with RPA, is a well-known diagnostic method that utilizes a fluorophore quencher (FQ)-labeled reporter gene to monitor the trans-cleavage activity of Cas12a [48,60]. In the presence of pathogens, the DNA substrate typically binds to the Cas12a-gRNA complex to change the protein conformation of Cas12a. The nearby FQ-labeled ssDNA reporter is non-specifically cleaved by Cas12a, resulting in the release of FQ and recovery of the reporter fluorescence. Several Cas12-based diagnostic tools have been employed to detect various viruses, particularly SARS-CoV-2, following the global COVID-19 pandemic that emerged in late 2019 (Table 3). Notably, this system can quickly respond to emerging infectious pathogens and has a critical role in controlling epidemics and disease spread. Here, we provide an overview of a highly representative Cas12a-based tool developed for the detection of SARS-CoV-2.
SARS-CoV-2 can be detected using an in vitro specific CRISPR-based assay for nucleic acid detection (iSCAN) [21], which combines a CRISPR-Cas12a detection system with the RT-LAMP technology. This novel detection platform, called two-pot iSCAN, has several benefits, including substantial time savings, high accuracy, enforceability, and ease of operation [21,41]. Most CRISPR-Cas12a-based detection tools, including iSCAN platform, typically involve two steps, which complicate the testing process and increase the risk of contamination [84]. POCT may require minimal liquid handling and “one-pot” reactions. Normally, this barrier can be overcome by mixing all reaction components into a single tube for a one-pot reaction [21,85]. The one-pot iSCAN system can be used for robust testing and identification of other pathogens and provides a new basis for the development of early pathogen detection platforms. The application of one-pot detection systems to the domain of self-inspection by non-technical personnel faces substantial hurdles due to difficulties in finding suitable materials and carriers to control the incorporation of the Cas12a/b-sgRNA complex into the reaction system. A technique recently developed by Broughton et al. [61] might be able to address this barrier.
The CRISPR-Cas12a diagnostic system relies on the indiscriminate cleavage of ssDNA by Cas12a activation; however, there are only two activation modes based on single- or double-stranded activators, which hinder specificity and universality [78]. Further, the Cas12a activation velocity affects the performance of the detection system. Therefore, new activation patterns have been developed to address these issues. A novel approach to detect Pseudomonas aeruginosa, developed by Sheng et al. [77], activates the CRISPR-Cas12a system by harnessing hydrazone chemistry, which has superior reactivity and selectivity [86,87], and improves the specificity and flexibility of the method.
To break through the technical bottleneck created by the presence of only two activation methods, Khodakov et al. [88] developed an unprecedented approach to Cas12 activation by using a toehold-mediated strand displacement reaction that strictly follows base-pairing rules. The toehold-mediated strand displacement reaction can also be applied for the detection of DNA point mutations such as that used in cancer diagnosis where DNA point mutations are a major cause of carcinogenesis and important markers of prognosis and diagnosis [89–91]. A combination of specific techniques, such as RPA, LAMP, rolling circle amplification [92], and primer exchange reaction (PER) [93] with the CRISPR-Cas technology, can promote the development of diagnostic platforms that use a one-step reaction as promising POCT tools [78,94].
Cas14, which was first reported in 2018 by the Doudna group [53], has several unique traits compared to Cas12 [46]. Cas14a has 530 amino acids, and is the smallest RNA-guided nuclease identified to date. Cas14 binds and cleaves ssDNA, and RNA is a central component of the Cas14a-sgRNA complex [37,46]. Cas14 does not require a target sequence, such as protospacer adjacent motif (PAM), for cleavage activity [95,96]. DETECTR-Cas14 is a recently developed diagnostic tool that combines an RPA tool with Cas14 and is based on the ability of Cas14 to cleave ssDNA through non-specific trans-cleavage activity [53]. This molecular scissors-based platform harnesses the ability of Cas14 to detect DNA with high fidelity to elicit the appearance of a fluorescent signal following the collateral cleavage of ssDNA [53]. This approach provides new possibilities for the detection of viruses [46,97]; however, the limited number of studies that have reported CRISPR-Cas14-based diagnostic platforms only elucidate the prospects of the application. The DETECTR-Cas14 platform makes it possible to study the effects of ssDNA on the human virome, which is an important component in understanding virus-disease associations [46,98]. The CRISPR-Cas14-based platform still requires nucleic acid extraction and amplification, a procedure that is undoubtedly cumbersome. A new approach that involves heating unextracted diagnostic samples to obliterate nucleases (HUDSON) in combination with CRISPR-Cas14 without nucleic acids extraction process and shipping of the samples simplifies the workflow [46,80].
Furthermore, this new molecular scissors-based diagnostic tool exhibits great potential in SNP identification [53]. The CRISPR-Cas14 system has been applied to molecular diagnostics and SNP genotyping. In addition, CRISPR-Cas14 coupled with metal elements has also been harnessed for the sensitive detection of non-nucleic acid targets [99,100]. There is no doubt that CRISPR-Cas14 detects ssDNA viruses and foodborne bacteria in clinical settings; however, the CRISPR-Cas14-based diagnostic platforms have been explored to a more limited extent than the CRISPR-Cas12-based platforms. Therefore, the applicability for non-diagnostic purposes remains to be investigated.
Diagnostic tools built on type VI CRISPR-Cas systems
CRISPR-Cas13-based platforms are promising diagnostic tools and potential next-generation diagnostic strategies for virus detection. Specific high-sensitivity enzymatic reporter unlocking (SHERLOCK) based on Cas13 was reported prior to the introduction of DETECTR. Nonetheless, both approaches are based on similar detection principles and require combination with isothermal amplification technologies [80]. The CRISPR-Cas13-based diagnostic platforms directly detect RNA viruses, such as ZIKV, DENV, HIV, SARS, and SARS-CoV-2, because Cas13 identifies RNA substrates rather than DNA substrates. Isothermal DNA amplification using RT-RPA also detects DNA viruses [80]. Furthermore, SHERLOCK has been shown to detect pathogenic bacteria, such as KPC and NDM-1. The next version of SHERLOCK (SHERLOCKv2) has been reported to provide a robust, 3.5-fold increase in sensitivity by coupling Cas13b to Csm6 [79].
Although SHERLOCK can specifically and precisely detect ZIKV and DENV at the level of 1 copy/μL [82], complicated operating procedures, especially nucleic acid extraction, have hindered widespread utility. HUDSON was developed to compensate for this deficiency and to simplify SHERLOCK by reducing lysed viral particles and deactivating RNases in body fluids [80,82]. HUDSON permits the use of RPA after HUDSON to directly detect virus from body fluids without the need for nucleic acid extraction, dilution, and purification [101]. A viral diagnostic platform that pairs SHERLOCK and HUDSON has been constructed to effectively and rapidly detect ZIKV and DENV [82]. This new platform is highly sensitive and specific and can be used without instruments. More importantly, it is a potential platform for multiplexing detection of viruses [79].
In 2020, Ackerman et al. [81] introduced combinatorial arrayed reactions for multiplexed evaluation of nucleic acids (CARMEN) as a flexible platform that enables highly multiplexed detection of pathogens. The combination of CARMEN with Cas13 (CARMEN-Cas13) stably detects >4500 crRNAs-substrate complexes in a single array. The CARMEN-Cas13 platform detects Middle East respiratory syndrome virus, SARS, SARS-CoV-2, HIV, ZIKV, DENV, hepatitis C virus, and HPV4, but not γHPV or Aroa virus. The flexibility and high throughput of CARMEN allow the introduction of crRNAs or amplification primers to detect emerging and re-emerging pathogens. Additionally, CARMEN complements the shortcomings of next-generation sequencing. In summary, the CARMEN-Cas13 platform expands the application of CRISPR-based platforms for pathogen detection by providing flexible testing.
RAPID DETECTION PLATFORMS BASED ON APTAMERS
CRISPR-Cas-based diagnostic platforms are cumbersome due to complex manipulations that are needed, such as extraction and amplification of nucleic acids. These tools have shown robust functionality in pathogen detection. Simple, convenient, and highly effective tools are urgently needed. Aptamer-based biosensor detection methods have subsequently attracted attention to address this issue.
Aptamers are 20–80-base ssDNA or RNA sequences identified by screening of nucleic acid libraries with the systematic evolution of ligands using an exponential enrichment (SELEX) strategy [102,103]. Aptamers are excellent alternatives to antibodies for antigen-specific identification [104]. Furthermore, aptamers permit stable immobilization of target molecules, such as viruses, bacteria, amino acids, proteins, and cells [105]. Typically, the screened aptamer binds specifically to the targeted substrate and indirectly detects pathogens based on physical, chemical, or electrochemical properties, such as changes in current or electrical resistance or the naked-eye recognition of fluorescence. Therefore, platforms based on aptamer-based biosensors (aptasensors) have been broadly used to detect pathogenic viruses and foodborne bacteria in clinical settings or diagnosis of human diseases (Fig 1). Aptasensors are divided into optical and electronic biosensors depending on the type of transducer.
Detection of bacteria via aptamer-based tools
Bacteria colonize a wide range of nature environments, and infection with pathogenic bacteria can cause severe clinical illness. Therefore, the early and rapid detection of bacteria in food or water samples is essential. Currently, most of the described aptamer-based detection tools are used for the detection of E. coli and S. aureus.
E. coli is a pathogenic microorganism widely found in nature and the human gut. E. coli serotypes can cause various clinical symptoms, such as diarrhea, abdominal pain, ulcers, and hemolysis [106,107]. S. aureus is a common pathogen that causes infectious diseases, such as pneumonia, arthritis, and sepsis [108]. In addition, following the abuse of antibiotics in clinical settings and the addition to poultry feed, S. aureus has gradually become insensitive to clinical treatment, a considerable challenge for human health [109]. Early and rapid diagnosis is very important in efforts to curb the spread of E. coli and S. aureus, guide medical treatment, and detect bacteria in food and water sources. Development of methods for the quantitative and rapid detection of E. coli and S. aureus has attracted the attention of food safety, medicine, and other fields, and fast and precise detection techniques based on aptamers have been exploited. The combination of optical or electrochemical sensors with aptamers has been widely used to test E. coli and S. aureus proteins, and even whole cells.
Aptamers assembled with optical sensors for the detection of E. coli and S. aureus
Visible light, ultraviolet light, and fluorescent sensors can be used to detect E. coli using aptamers; these approaches provide several advantages, including ease of operation and low cost compared to ELISA, which is a conventional technique that requires an abundant amount of antibodies and is expensive [110]. Several optical-sensors-based tools for detecting E. coli have already been developed. In 2016, Fu et al. [111] developed a method based on sandwich-like assay to detect E. coli K88. The authors introduced biotin-labeled and Au nanoparticle (NP)-labeled aptamers to create a sandwich-type complex (biotin-labeled aptamer I/E. coli K88/NP-labeled aptamer II). The presence of E. coli K88 was visualized based on a color reaction, and the assay had a linear range of 10–105 colony-forming units (CFU)/mL. A colorimetric method based on an aptamer-polydiacetylene optical sensor was reported to detect E. coli O157:H7 at ranges between 104 and 108 CFU/mL [112]. Pandit et al. [113] developed a new method to detect E. coli in water samples. Specifically, two E. coli-specific aptamers, which were identified by screening, were separately conjugated with superparamagnetic iron oxide NPs and CdTe-MPA quantum dots, respectively. The use of CdTe-MPA quantum dots allows the qualitative and quantitative estimation of E. coli by fluorescence spectrophotometry. The limit of detection (LOD) of this biosensor is as high as 102 CFU. Yuan et al. [114] developed a gold NP-based colorimetric aptasensor to detect S. aureus based on tyramine signal amplification technology. In this method, the biotinylated anti-S. aureus aptamer is immobilized on the surface of a microtiter plate before the addition of other necessary ingredients (biotin-tyramine, streptavidin-horseradish peroxidase, avidin-catalase, and S. aureus). The catalase added during the assay depletes endogenous hydrogen peroxide. Finally, gold (III) chloride trihydrate is used to change the color of the reaction product and the presence of S. aureus is determined by measuring absorbance at 550 nm. The assay could detect S. aureus ranging from 10-106 CFU/mL under ideal conditions, and the LOD was 9 CFU/mL. Aptamer-based techniques using fluorescence for the detection of E. coli and S. aureus exhibit higher sensitivity than techniques based on visual and colorimetric assessment. Fluorescence aptamer-based techniques often require sophisticated instruments, such as epifluorescence, confocal fluorescence, and confocal laser scanning microscopy.
Aptamers assembled with electrochemical sensors for the detection of E. coli and S. aureus
Electrochemical signals can be displayed by current, resistance, or potential, offering several possibilities for constructing electrochemical biosensors for E. coli and S. aureus testing [110]. Notably, aptamers coupled to electrochemical sensor detection platforms have the potential to achieve tractability and miniaturization and have been reported to directly detect pathogens in turbid media. A study recently described a method to detect the outer membrane proteins (OMPs) of E. coli through a change in resistance [115]. In that assay, potassium ferricyanide was used as a redox probe for electron exchange between the gold electrodes. E. coli OMPs combined with aptamers could hinder this process, leading to an electrochemical reaction. The relationship between the concentration of E. coli OMPs (1 × 10−7 – 2 × 10−6 mol/L) and electron transfer resistance was excellent. In addition to OMPs, E. coli lipopolysaccharide can be used as a detection site [116]. Furthermore, aptamer-based electrochemical sensors can be used to detect E. coli genes. For example, E. coli ribosomal RNA was detected by monitoring the oxidation state of guanine nucleotides [117], and E. coli genomic DNA was directly recognized by utilizing methylene blue as a DNA hybridization probe molecule [118]. Several tools have emerged for detecting whole cells of E. coli [119–121].
Nguyen et al. [122] reported a new method for the detection of S. aureus. Nguyen et al. [122] utilized a cognate pair of aptamers for S. aureus to fabricate a sandwich-type aptasensor (aptamer I/S. aureus/horseradish peroxidase-conjugated aptamer II) on a screen-printed gold electrode. S. aureus can be detected based on a significant decrease in faradaic peak current and peak-to-peak potential separation. The predicted LOD of this sandwich-type signal-on electrochemical biosensor was 39 CFU in buffer and 414 CFU in commercially available bottled water samples. Further, the new biosensor could easily detect S. aureus in a sample volume as low as 5 μL. As rapid, sample, sensitive, and specific approaches, sandwich-type electrochemical aptasensors have promising applicability in food safety management.
Detecting viruses via aptamer-based tools
Aptamer-based biosensors have been developed for bacterial and viral detection. Table 5 summarizes the aptasensor-based techniques for the detection of viruses. Optical aptasensors for virus detection can be categorized into six groups. Surface plasmon resonance (SPR) aptasensors are based on the detection of resonance of free electrons based on the change in refractive index. Aptamers are prefixed to the surface of a gold plate, and the thickness of the gold surface is altered after the capture of the virus; the presence of the virus is determined by measuring changes in the angle or the intensity of polarized light. Colorimetric aptasensors determine the presence of virus based on an apparent shift in color, which can be directly observed with the naked eye or using a spectrophotometer. Fluorescence aptasensors are based on the transition of fluorescence signals or the production of fluorescence polarization. Chemiluminescence (CL) aptasensors are commonly and extensively used to detect viruses, such as hepatitis B virus, HIV, SARS-CoV-2, and other emerging and re-emerging infectious viruses in clinical practice. These assays are based on colorimetric detection. The aptamer is co-incubated with virus particles, followed by the addition of catalytically-active complexes that bind to the virus, and the addition of a chromogenic or CL reagent for the quantitative detection of the virus. Surface-enhanced Raman scattering aptasensors utilize Raman scattering spectroscopy, which reflects the characteristics of the medium by the change in the energy of photons after passing a laser light through the medium. Interferometric aptasensors are the sixth category developed on the basis of interferometry. It is a label-free technique for measuring light intensity, which includes a refractive index or physical property.
Aptasensors applied in virus detection.
| Detection | Technique | Virus | Limit of Detection | Refs. |
|---|---|---|---|---|
| Optical aptasensors | SPR | (AIV) H5N1 | 0.128 HAU | [123] |
| 200 EID50/mL | [124] | |||
| HIV-1 | 0.12 ppm | [125] | ||
| Colorimetric | MNV | 200 MNV/mL | [126] | |
| H3N2 | 11.16 μg/mL | [127] | ||
| H5N1 | 0.1 μg/well | [128] | ||
| H5N2 | 1.27 × 105 EID50/mL | [129] | ||
| HCV | 11 nM | [130] | ||
| 3.91 × 102 FFU/mL | [131] | |||
| HuNoV | 10 RNA copies | [132] | ||
| ZIKA virus | 0.1 ng/mL | [133] | ||
| Multiplex strain-specific influenza virus | 2 × 106 virus particles | [134] | ||
| SFTS virus | 9 pg/mL | [135] | ||
| SARS-CoV-2 | 10 ng/mL | [136] | ||
| 1 ng/mL | [137] | |||
| Fluorescence | HBV | 1.25 mIU/mL | [138] | |
| SARS-CoV-2 | 130 fg/mL | [139] | ||
| RSV | 80 nm | [140] | ||
| H1N1 | 3.45 nM | [141] | ||
| CL | SARS-CoV | 2 pg/mL | [142] | |
| HBV | 0.1 ng/mL | [143] | ||
| SERS | H1N1 | 97 PFU/mL | [144] | |
| Interferometry | HCV | 700 pg/mL | [145] | |
| Electronic aptasensors | Electrochemical aptasensors | H1N1 | 10 pM | [146] |
| HCV | 0.16 fg/mL | [147] | ||
| 3.3 pg/mL | [148] | |||
| H5N1 | 8 × 10−4 HAU/200 μL | [149] | ||
| (AIV) H5N1 | 2−9 HAU | [150] | ||
| HIV-1 | 1 nM | [151] | ||
| SARS-CoV-2 | 1.6 × 10 PFU/mL | [152] | ||
| Noro viruses | 180 virus particles | [153] | ||
| Piezoelectric transducers | HIV-1 | 0.25 mg/L | [154] | |
| (AIV) H5N1 | 0.0128 HAU | [155] | ||
| HCV | 0.1 pM | [156] |
Furthermore, viruses can be detected with electronic biosensors, which are a type of electrochemical aptasensor, or piezoelectric transducers based on different mechanisms. Electrochemical aptasensors follow the principle of a change in electrical signal caused by the binding of the aptamer to the target virus. Aptamers used to capture viral molecules are prefixed on electrode plates. Detection tools based on electrochemical aptasensors can be further categorized into three subclasses: enzymatically-labeled aptasensors, in which enzyme catalysis triggers a change in the electrical signals; enzyme-free aptasensors, in which the binding of viral particles to the aptasensors directly causes a change in the electrical signal; and field-effect transistor (FET)-based aptasensors. The aptamer is immobilized on the FET surface to capture target viral molecules and the change in the charge distribution on the FET surface is detected and translated into an easy-to-read signal in FET-based aptasensor detection tools. Several viral assays based on these technologies have been developed (Table 5). Piezoelectric transducers are based on the piezoelectric effect principle, in which an electrical charge is generated when certain materials are subjected to mechanical stress.
Detecting parasites via aptamer-based tools
Parasites are a large family of pathogens that cause zoonoses. Seven of the top 10 global tropical diseases are caused by parasites. As such, parasites are a serious threat to human health and a significant burden to the economy, both of which can be mitigated by the rapid detection of parasites. Recently developed aptamer-based tools for parasite detection provide rapid and accurate approaches with a low LOD and have been widely implemented to identify and monitor various parasites.
Roca et al. [157] selected a DNA aptamer (D10) against Plasmodium falciparum 1-deoxy-D-xylulose-5-phosphate reductoisomerase (DXR) using the SELEX strategy, a vital enzyme in the methyl erythritol phosphate (MEP) pathway of isoprenoid biosynthesis. The D10 aptamer binds to recombinant DXR in vitro and explicitly targets the apicoplast, in which the MEP pathway is localized, an essential organelle in P. falciparum. Therefore, D10 is a highly specific marker for naive erythrocytes and red blood cells parasitized by P. falciparum and for the apicoplast. Therefore, the D10 aptamer is a promising lead for future diagnostic strategies for Plasmodium and a potential tool for other studies of apicomplexan parasites, such as Toxoplasma gondii. In addition, the D10 aptamer detects bacteria harboring the MEP pathway, such as E. coli and P. aeruginosa. To address the lack of aptamer-based methods for the detection of SAG-1, the major T. gondii surface protein, Cui et al. [158] developed a direct enzyme-linked aptamer assay to target native SAG-1 (nSAG-1) for the detection of T. gondii. This novel technique is a potential tool for the rapid and precise diagnosis of toxoplasmosis. Homann and Goringer [159] was the first to employ the SELEX strategy to screen aptamers for the detection of African trypanosomes. The authors identified an RNA aptamer that crosslinked to a 42-kDa protein in the flagellar capsule of trypanosomes through 13 rounds of SELEX selection. Interestingly, although aptamer-based assays have been proposed for the detection of parasites, with many tools developed over the years, only a handful of reports use whole cells as detection targets. Commonly, the screening substrates of aptamers for parasites are enzymes, proteins, and infected cells [160–162] and these aptamers detect the secretions and excretions of the target parasite or specific virulence-related molecules.
RAPID DETECTION PLATFORMS COUPLING CRISPR-CAS- AND APTAMER-BASED SYSTEMS
CRISPR-Cas- and aptamer-based techniques utilize many tools for pathogen detection and exhibit robust efficacy, sensitivity, specificity, and convenience compared to conventional detection approaches; however, cumbersome procedures and the detection of the LOD are issues that remain to be resolved. For example, CRISPR-Cas-based tools require nucleic acid extraction and amplification. Conversely, multiple rounds of aptamer screening are essential for aptamer-based platforms and detection with aptamers only does not achieve good sensitivity and the LOD is usually in the picomolar and micromolar ranges [3,163]. A study recently reported that these two complementary techniques can be combined to develop effective detection tools for pathogen detection to address these obstacles (Fig 2). Notably, this cross-cutting approach has been used to detect pathogenic bacteria in samples, and should be studied more for the diagnosis of viruses or parasites, and therefore has promising applications in the POCT of foodborne bacteria. This CRISPR-Cas- and aptamer-based detection platform plays an essential role in preventing the spread of pathogens in resource-poor countries and regions. Some of the complementary tools can distinguish between dead and living bacteria, providing a feasible method for further control of pathogenic bacteria at the source.
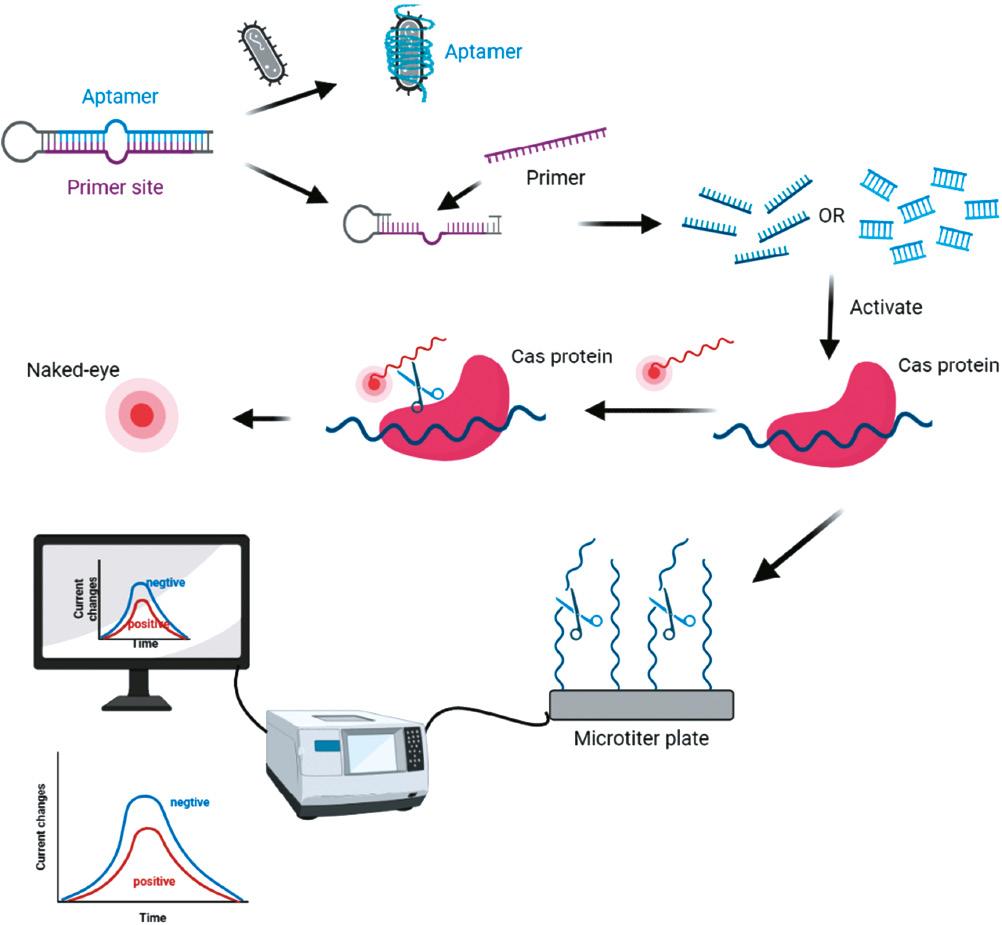
Schematic illustration of CRISPR-Cas systems coupled with aptamers for pathogen detection. Aptamers are usually prestabilized in various ways. In the presence of the target pathogen, the aptamer dissociates from the system and the remaining components synthesize a short-chain nucleic acid to activate the Cas protein which then cleavages the indicated probe, leading to a change in fluorescence or current.
Diagnostic tools built on the CRISPR-Cas12 system coupled with aptamers
Bu et al. [164] constructed an electrochemical aptasensor to detect pathogenic bacteria without nucleic acid extraction by combining the CRISPR-Cas12a system with PER. In this approach, the DNA aptamer binds to the hairpin loop of the primer, which remains inactive in the absence of pathogens, thereby hindering primer amplification. In the presence of target bacteria, the DNA aptamer disentangles the hairpin loop structure and the primer is extended to form ssDNA, which activates the trans-cleavage activity of CRISPR-Cas12a to crush the MB probe pre-immobilized on the Au surface, resulting in a decrease in the monitored peak current. This novel technique has been utilized to detect bacteria in milk samples, such as the detection of E. coli O157:H7 with an LOD of 19 CFU/mL. To expand the repertoire of diagnostic methods for various disease biomarkers, Yuan et al. [165] recently developed a “signal on-off-super on” sandwich-type aptasensor of CRISPR-Cas12a coupled voltage enrichment for the detection of vascular endothelial growth factor with an LOD of 0.33 pM. Using this technique, AuNPs@Ti3C2Tx-Mxene was synthesized by immobilizing AuNPs on the surface of Ti3C2Tx-Mxene, which stabilized the trapped aptamer and therefore altered the electrochemical signal more effectively. This approach enriched ssDNA using an electrochemical signal via CRISPR-Cas12a-coupled voltage enrichment instead of amplification.
Diagnostic tools built on the CRISPR-Cas13 system coupled with aptamers
Many platforms combining CRISPR-Cas13 with aptamers have been developed for pathogen detection. The difference between these platforms and the CRISPR-Cas12a-based platforms lies in the Cas enzyme. Shen et al. [55] presented the new APC-Cas platform for the detection of S. enteritidis. The allosteric probe used in this platform contains three sections: the T7 promoter; an aptamer; and a primer-binding site. The probe is in a quiescent state due to the integration of the primer-binding site into the aptamer. In the presence of the target pathogen, the allosteric probe undergoes conformational changes and ssRNA is generated using the T7 RNA polymerase. The platform exhibits strong sensitivity and functionality for the quantitative detection of S. enteritidis and is a promising approach for pathogen detection. Zhang et al. [25] reported a system to distinguish between viable and dead pathogenic bacteria using a light-up RNA aptamer combined with CRISPR-Cas13; the LOD of this system was 10 CFU for the detection of Bacillus cereus.
Diagnostic tools built on the CRISPR-Cas14 system coupled with aptamers
Cas14 is the smallest identified Cas enzyme and does not require PAM sequence identification; therefore, CRISPR-Cas14-based aptasensors exhibit enhanced sensitivity and versatility compared to the biosensors based on Cas12 or Cas13. Although reports of Cas14-based aptasensors are rare, Cas14-based aptasensors may be more potent. AcasB, a novel platform developed for on-site detection of live S. aureus, contains an aptamer biosensor combined with Cas14a1 [3]. This platform includes 3 probes, a 45-nucleotide aptamer to target live S. aureus, a 21-nucleotide blocker for activation of Cas14a1, and an sgRNA to bind the blocker. Importantly, this platform does not require nucleic acid extraction and amplification. The blocker bound to the aptamer forms a stable hybrid, which dissociates to release the blocker in the presence of live S. aureus and specifically targets the aptamer. The subsequently released blocker activates Cas14a1 by binding to sgRNA, which then cleaves the surrounding fluorescence probe, resulting in fluorescence. This platform directly distinguishes whether the bacteria are alive or dead and has an LOD of 400 CFU/mL for live S. aureus in the tested samples.
Cas14 combined with aptamers can also be used to detect non-nucleic-acids targets. Zhou et al. [166] developed a biosensor platform designated as a highly-sensitive aptamer-regulated Cas14 Rloop for bioanalysis (HARRY). A diblock ssDNA was designed to accommodate the aptamer and Cas14 activation sequences in this platform. The ssDNA activates Cas14a, then the Cas14a trans-cleavages the fluorescent reporter, causing fluorescence enhancement in the absence of the target. Non-nucleic acids substrates, such as ATP, Cd2+, histamine, aflatoxin B1, and thrombin, have been detected by the HARRY platform, which possesses LOD at the nanomolar level.
CONCLUSIONS AND FUTURE PERSPECTIVES
Pathogens pose a severe threat to the survival of humans and animals, and are a public health issue of concern. CRISPR-Cas- and aptamer-based systems have been harnessed for POCT research primarily based on the application of CRISPR-Cas systems for pathogen detection. Conventional diagnostic methods are complicated and cumbersome. Therefore, diagnostic platforms based on the CRISPR-Cas system and aptamers have been exploited to improve detection efficiency and simplify the procedures, while reducing cost and improving applicability. Nevertheless, most of these tools are theoretical and few have been introduced for commercial use. The reasons that prevent the marketization of these tools, i.e., nucleic acid extraction and amplification, are a necessary component of CRISPR-Cas-based approaches, which are cumbersome processes that require skill. Notably, technological advances, such as isothermal amplification, microfluidics, and gene chips, should overcome these barriers [78]. SELEX is a time-consuming, not-yet-mature, costly, and unstable strategy, and sensitive simultaneous detection of multiple pathogens in a mixed medium is challenging. For clinical samples, the selected aptamers cannot be completely and accurately matched, and the affinity and specificity change [29]. Recent studies achieved the detection of pathogens in turbid media and the optimization of the SELEX screening process. Consequently, combination of the complementary CRISPR-Cas- and aptamer-based systems is an effective optimization approach to fill the gaps in pathogen detection. Indeed, the combination of these two systems has gradually replaced the traditional detection methods over the past decade. However, combinations of various Cas proteins with different aptamer techniques, such as allosteric probe-initiated catalysis and PER, are typically limited to bacterial detection, therefore, future studies should be conducted to advance viruses and parasite detection in clinical settings.
The recently described CRISPR FISHer [167] can potentially track particular cell-intrinsic or exogenous DNA sequences, broadening the application range of live cell imaging and laying the foundation for biomedical diagnostics. Several recent studies have identified new Cas proteins in nature. For instance, Cas12a2 [168,169] is activated upon binding to RNA targets and can efficiently degrade ssRNA, ssDNA, and dsDNA. In contrast to the previously described Cas proteins, Cas12a2 is programable and capable of pathogen detection. Cas14 is a newly discovered and promising protein for the development of more efficient and convenient detection tools, while its use in conjunction with aptamer-based systems for the detection of non-nucleic acids offers a new idea for broadening diagnostic targets and establishing detection methods. Many aspects of the mechanism underlying the Cas protein effect, both newly and previously discovered, are still not fully understood. In addition, combining several Cas proteins with different functions to enable simultaneous pathogen detection and treatment is an intriguing potential approach.
In conclusion, pathogen detection tools should be practical and generalizable, ideally with test results linked to a smartphone or smart bracelet, enabling citizens to self-test for pathogens in food or water and alleviating stress. With the constant expansion of technology, there is great promise for improvement in diagnostics, and there is an urgent need to establish novel detection tools to respond rapidly to emerging zoonotic pathogens in the near future.

