
Introduction
Hepatocellular carcinoma (HCC), which ranks fourth in deaths caused by cancer [1], causes over 600,000 deaths annually worldwide [2]. Over the past few years, considerable progress has been made in the epidemiology, molecular profiles, and risk factors for HCC. However, the 5-year survival rates of HCC patients in China are still less than 12% because many patients are not diagnosed until the cancer has developed to an advanced stage [3, 4]. In addition to the high molecular heterogeneity of HCC at three levels, namely interpatient, intertumoral, and intratumoral [5], it is imperative to explore the underlying molecular mechanism of HCC to identify novel effective prognostic factors that can be applied to diagnose HCC patients at an early stage and to reduce the mortality of HCC patients.
N6-Methyladenosine (m6A) regulates RNA metabolism, including splicing, export, degradation, and microRNA processing, through m6A methyltransferases (writer; the enzymes that deposit modification), demethylases (eraser; the enzymes that remove modification), and reader proteins (reader; the proteins that bind with the m6A sites of targeted RNA) [6–9]. The m6A modification is methylated at the N6 position on adenosine, which affects the localization, translocation efficiency, and variable splicing of mRNA [10]. It has been confirmed that m6A modification participates in regulating gene expression and plays important roles in many biological processes. Previous studies have reported the complex mechanisms of reversible m6A modification in human diseases [6, 11–13], even in cancer. Dynamic m6A levels have been demonstrated to participate in tumor development by modulating the expression of tumor-related genes in different types of tumor [7].
Recently, there is increasing evidence that dysregulation of m6A regulators may be involved in the progression of cancer, influencing the prognosis for patients with cancer. For instance, previous studies have shown that m6A methyltransferases, such as METTL14, are highly expressed and promote leukemogenesis in regulating targeted mRNAs through m6A modification in acute myeloid leukemia [14] as well as suppressing tumor progression by regulating the expression of related genes in a m6A modification manner [15]. In addition, YTH domain family member 2 (YTHDF2), a member of the YTHD family, suppresses the proliferation of HCC cells by destabilizing targeted mRNA [16]. These studies indicate that m6A serves important roles in the development of HCC. However, the association between m6A-related genes and HCC prognosis remains unclear.
In the present study, we obtained the RNA transcriptome and clinical data of HCC from The Cancer Genome Atlas (TCGA). We found three m6A regulators and 192 candidate m6A-related genes that were differentially expressed in different stages of HCC. We then analyzed the relationship between the expression of m6A candidate genes and progression in HCC patients. We finally selected 19 m6A-related genes to construct a risk model based on the risk score and explored the function of m6A-related genes in HCC development and prognosis. Our results provide new ideas for the integrated roles of m6A regulators and m6A-related genes in HCC.
Materials and methods
Data collection
RNA-seq and clinical data
We performed an analysis on TCGA level 3 RNA-seq transcriptome data. The FPKM data of HCC RNA-seq were downloaded from TCGA database along with the clinical data of the corresponding samples. In addition, we downloaded gene annotation files from Genecode. We selected samples that had both pathological characteristics (including stages I, II, III, and IV) and RNA-seq expression data. The epidemiological characteristics of HCC patients are presented in Table 1 . We also downloaded the FPKM data for the HCC RNA-seq and the clinical data for the LIHC samples from the International Cancer Genome Consortium (ICGC) database.
Clinicopathological Characteristics of LIHC Patients (n = 374) from TCGA Database
| Age | |
| ≤60 years | 260 (69.52) |
| >60 years | 113 (30.21) |
| Not reported | 1 (0.27) |
| Sex | |
| Female | 121 (32.35) |
| Male | 253 (67.65) |
| Pathological stage | |
| Not reported | 24 (6.42) |
| Stage I | 173 (46.26) |
| Stage II | 87 (23.26) |
| Stage III | 85 (22.73) |
| Stage IV | 5 (1.34) |
| Survival time | |
| ≤3 years | 105 (28.07) |
| >3 years | 25 (6.68) |
| Alive or not reported | 244 (65.24) |
| Survival status | |
| Alive | 243 (64.97) |
| Dead | 130 (34.76) |
| Not reported | 1 (0.27) |
Values are presented as n (%).
Integration analysis of differentially expressed m6A-related genes in HCC
After selecting liver cancer samples that had both pathological characteristics and RNA-seq data, we applied the gene annotation file to construct a profile of m6A-related genes in liver cancer (FPKM data). According to the stage standard for LIHC, these liver cancer samples were divided into 4 groups: stage I, stage II, stage III, and stage IV. We compared the expression values of the ICGC and TCGA data between different stages of HCC using one-way analysis of variance (p < 0.05). And then the selected differentially expressed genes were further used as the m6A candidate gene sets. Finally, a heatmap was constructed to visualize the differences.
Consensus clustering analysis
To elucidate the biological characteristics of m6A regulators in LIHC, we used the “ConsensusClusterPlus” package in R to perform an unsupervised cluster analysis on the samples [20]. All tumor samples corresponding to the candidate m6A-related genes were classified into two subgroups: KM1 and KM2.
Integration analysis of pathological characteristics and survival time of the KM1 and KM2 subgroups
We used a t-test to analyze the distribution of age between the KM1 and KM2 subgroups. In addition, we analyzed the difference in the World Health Organization (WHO) classification via the chi-square test and utilized the Cox regression model to explore the survival time difference between the KM1 and KM2 subgroups.
Interaction analysis among m6A candidate gene sets in HCC
The STRING database was used to explore the protein–protein interactions (PPIs) among m6A candidate genes. We used Spearman’s correlation analysis to explore the relationship among different m6A candidate genes.
Identification and enrichment analysis of differentially expressed genes between the KM1 and KM2 subgroups
After constructing the m6A candidate gene expression profiles of KM1 and KM2, we performed principal component analysis (PCA) to analyze the differentially expressed genes. We then used DAVID 6.759 to conduct functional annotation and enrichment analysis for the differentially expressed genes.
COX regression analysis and calculation
To determine the prognostic value of m6A candidate genes in HCC, a risk signature was developed for the dataset using univariate Cox regression analysis. Next, we filtered several important factor genes as prognostic genes of liver cancer. The risk score was calculated using the following equation:

where Coef i is the coefficient and Exp i is the expression of prognostic genes in each liver cancer sample. According to the median risk score, we divided the samples into low- and high-risk groups.
Risk scoring for prognosis and pathological characteristic prediction
We applied the LASSO regression algorithm (“glmnet” package in R) to TCGA liver cancer samples and obtained the risk score of 98 prognostic genes in each TCGA liver cancer sample. The 98 genes were then used to establish risk characteristics (LASSO automatically selected the best genes as characteristics) and the 1- to 5-year survival outcome of the HCC patients, and the grouping results for KM1 and KM2 were used as the predicted outcome. The predictive efficiency was then estimated by receiving operator characteristic (ROC) curves using the R software “survival” package.
Similarly, we applied the LASSO regression algorithm (“glmnet” package in R) to the ICGC liver cancer samples and obtained the risk score of 98 prognostic genes in each ICGC liver cancer sample. The 98 genes were then used to establish risk characteristics (LASSO automatically selected the best genes as characteristics), and the prognostic outcome was used as the predicted outcome. The predictive efficiency was then estimated by ROC curves with the “survival” package in R. The results indicated that the risk score provided predictors of prognostic outcome for cancer patients.
Analysis of differences in immune infiltration between low- and high-risk score groups
CIBERSORT provided a method to characterize cell composition based on gene expression profiles of complex tissues. The “LM22” white blood cell characteristic gene matrix composed of 547 genes was used to distinguish 22 immune cell types. In the present study, we used the results of the CIBERSORT evaluation published by Thorsson et al. [21]. We used the proportion of immune cells in samples from the low- and high-risk groups of HCC for statistical testing to analyze the difference in immune infiltration between the two subgroups.
Integration analysis of mutation differences and correlation analysis of risk score with matrix, immune score, and mRNAsi dryness index
We downloaded the liver cancer SNV data from TCGA and used the “maftools” package in R to display the distribution of mutations in the two subgroups. Based on the expression profile, we used the “estimate” package in R to evaluate the matrix score and immune score of the samples. According to the results of the tumor stemness index published by Malta et al. [22], the stemness index of liver cancer was extracted. The correlation of the immune score, stromal score, or mRNAsi with the two risk groups was then evaluated based on Pearson’s correlation coefficient analysis.
Results
Identification of m6A methylation candidate genes in HCC
We obtained liver cancer samples that included both pathological characteristics and RNA-seq expression data. Based on the liver cancer stage, these samples were divided into 4 subgroups: stage I, stage II, stage III, and stage IV. After overlapping 1258 m6A-related genes of LIHC, we obtained differentially expressed genes (p < 0.05) among the four sample types by variance analysis. Finally, 192 differentially expressed genes were screened out as the m6A candidate gene set ( Table S2 ). Notably, three of the m6A regulators, YTH domain-containing 1 (YTHDC1), YTH N6-methyladenosine RNA-binding protein 1 (YTHDF1), and YTH N6-methyladenosine RNA-binding protein 2 (YTHDF2), were expressed differently in the four subgroups. We then used the 192 m6A candidate genes to construct an expression heatmap. As shown in Figure 1 , there was a significant difference among the expression levels of m6A candidate genes in different stages of HCC.

Consensus clustering analysis
We performed consistent cluster analysis of HCC patients according to the expression of m6A regulators from TCGA. To explore the roles of m6A candidate genes in HCC, we used the expression levels of the 192 m6A candidate genes as feature vectors, and we then divided the HCC samples into different subgroups using the “Consensus Cluster Plus” package in R. In a clustering range from 2 to 10, we obtained a relatively clustering stable optimal k value (2) with TCGA data. The specific grouping conditions are presented in Table S3 , and the conditions of sample-consistent clustering are shown in Figure 2 . The consistent cluster cumulative distribution function (CDF) for k = 2–10 is presented in Figure 2A . The relative change in the area under the CDF curve is presented in Figure 2B ; meanwhile, Figure 2C shows the visualization of the consistent clustering matrix between groups at k = 2. The distribution of the sample when k was between 2 and 10 is shown in Figure 2D . We named the subgroups KM1 and KM2.
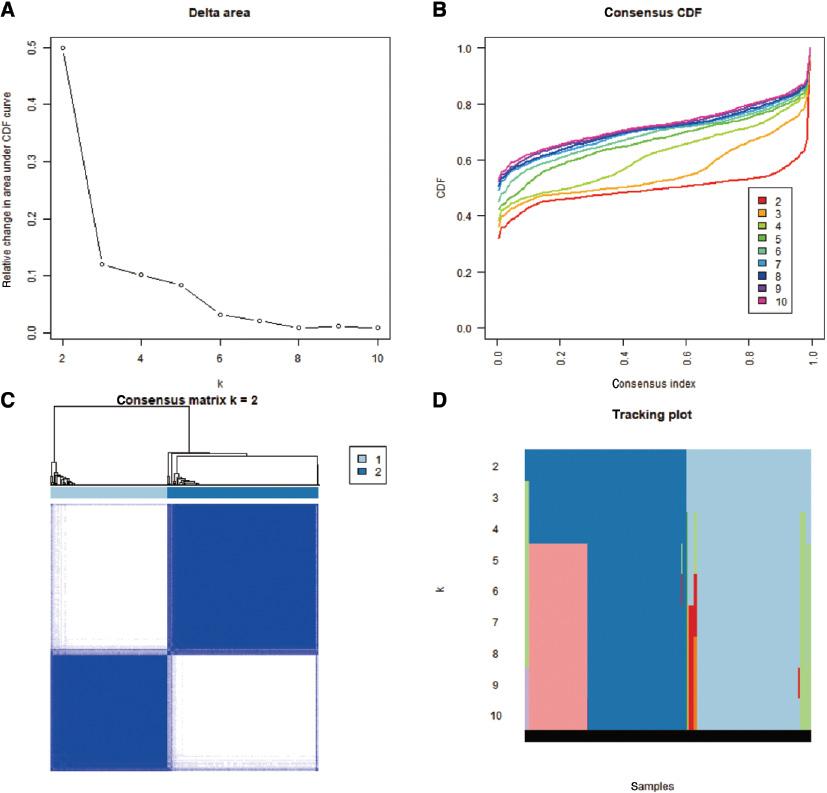
Consistent cluster analysis of hepatocellular carcinoma. (A) The consistent cluster cumulative distribution function (CDF) when k is between 2 and 10. (B) The relative change in the area under the CDF curve when k ranges from 2 to 10. (C) Visualization of the consistent clustering matrix between groups at k = 2. (D) The distribution of the sample when k is between 2 and 10.
Integration analysis of pathological characteristics and survival time of the KM1 and KM2 subgroups
We extracted the clinical information from the two subgroups ( Table S4 ) and performed a t-test to analyze the age distribution of the KM1 and KM2 subgroups. There was a significant difference in age between the two subgroups (p = 0.004) ( Figure 3A ). We extracted the samples containing stage data and analyzed the differences in the WHO staging of the two subgroups via the chi-square test. The WHO classifications were significantly different between the two subgroups (p < 0.001). The WHO classification pie chart of the two subgroups is shown in Figure 3B . We also extracted samples containing survival time from the KM1 and KM2 subgroups, and Cox regression was then used to explore the survival time between the two subgroups. Figure 3C shows the KM survival curve, and the results indicated a significant difference (p = 0.004) in survival time between the two subgroups.

Comparative analysis of pathological characteristics and survival time between the two subgroups. (A) Box plot of age between the two subgroups of liver cancer. There was a significant difference in age (p < 0.05). (B) WHO classification pie chart between two subgroups of liver cancer. There was a significant difference in WHO classification (p < 0.001). (C) Survival curves of the two subgroups in liver cancer. There was a significant difference in survival time (p < 0.01).
Interaction analysis among m6A candidate gene sets in HCC
To explore the association network of functional proteins among these 192 m6A candidate genes, we generated a PPI network of the candidate gene set using STRING ( Figure 4A ). We then calculated the Spearman’s correlation between every two m6A candidate genes based on the m6A candidate gene expression profile. In this study, we selected genes with cor > 0.6 and p < 0.05 ( Table S5 ) to construct a correlation network ( Figure 4B ). Spearman’s analysis was used to analyze the correlation of the expression levels among the factors. The correlation coefficient threshold of the expression level was |r| ≥ 0.3, and the p value of the rank sum test was p < 0.01. As shown in Figure 4C , the correlation of all 192 genes was analyzed. In Figure 4C , blue represents a positive correlation, whereas red represents a negative correlation.

Interaction analysis among m6A candidate gene sets in hepatocellular carcinoma. (A) Protein-protein interaction (PPI) network of m6A candidate genes. (B) The coexpression network of m6A candidate genes. (C) Correlation of expression from 192 liver cancer-related genes. Blue represents a positive correlation, and red represents a negative correlation.
Identification and enrichment analysis of differentially expressed genes between KM1 and KM2
We explored the expression differences of samples between the two subgroups via PCA. The results ( Figure 5A and B ) suggested that the RNA expression of the two subgroups was specific. However, there were still some overlapping areas in the two subgroups, suggesting that both subgroups may share some commonality.

Identification and functional enrichment analysis of differentially expressed genes between KM1 and KM2. (A) Principal component change range. (B) Principal component analysis of the KM1 and KM2 subgroups. (C) GO functional enrichment of differentially expressed genes between the KM1 and KM2 subgroups. (D) KEGG functional enrichment of differentially expressed genes between the KM1 and KM2 subgroups.
We then used a t-test to explore the differentially expressed genes between the two subgroups (p < 0.05) ( Table S6 ). GO function and KEGG pathway enrichment analyses of the differentially expressed genes were performed with DAVID ( Table S7 and Table S8 ). The results indicated that the differences between the two subgroups were mainly enriched in several GO functions, such as cell membrane-related lipids and proteins as well as intracellular energy protein binding. These genes were also enriched in pathways associated with Fanconi anemia and homologous recombination. These pathways were closely related to DNA repair, cross-linking, and precise repair of DNA double-strand breaks ( Figure 5C and D ).
Analysis of univariate Cox regression and prognostic value of m6A candidate genes
To explore the prognostic value of m6A candidate genes in HCC, we used the “survival” package in R to perform univariate Cox regression analysis. A total of 343 samples remained after removing those samples without survival-related information. The results indicated that among 192 candidate genes, the expression of 98 candidate genes correlated with the survival of HCC (p < 0.05).
To further predict the prognosis of m6A regulatory genes in liver cancer, we analyzed 98 gene sets with the LASSO regression analysis algorithm. As a result, 19 genes were obtained to construct risk characteristics. Of these 19 genes for the prognostic model, POMT2, DEPDC1, UNG, GEMIN5, OPRM1, THADA, TET1, BBS7, TBCD, DNAAF5, INTS1, BRSK2, RABIF, COPB2, ZMYM1, COPS8, and ZNF337.AS1, were related to poor overall survival, whereas regucalcin (RGN) and cysteine sulfinic acid decarboxylase (CSAD) were related to good overall survival in LIHC ( Figure 6A–C ). We then used the regression coefficient to calculate risk scores. To clarify the prognostic efficacy of these characteristics, we divided the samples into two subgroups according to the median value of risk scores. As shown in Figure 6D , the high- and low-risk groups had significant differences in survival (p < 0.001). To validate the stability of the risk scores, we utilized the ICGC data to evaluate the efficacy of these features. The results suggested that 17 of the 19 candidate genes with significant survival existed in the ICGC data, and the remaining two genes had no expression (as indicated by “NA”). We used the expression of the 17 genes for survival analysis, which indicated that there was no significant difference in the survival results (p = 0.411) ( Figure 6E ).

Nineteen survival-related m6A RNA methylation-related genes were screened. (A) Least absolute shrinkage and selection operator (LASSO) regression analysis was used to screen 98 m6A RNA methylation-related genes. (B, C) The 95% confidence intervals (CIs) obtained by univariate Cox regression and the coefficients calculated with multivariate Cox regression using funnel plots. (D, E) Kaplan-Meier curves of the two groups from TCGA and ICGC.
Prognostic value of m6A candidate genes with different clinicopathological features and clinical outcomes for HCC
We analyzed the differences in age, sex and stage of the patients in the two subgroups. The results showed that sex (p = 0.008) and stage (p < 0.001) in the high-risk group were significantly different from those in the low-risk group, whereas there was no obvious difference in age between the two groups (p = 0.178). We also explored the correlation between risk scores and different clinical characteristics. The results indicated significant correlations between risk score and sex (p = 0.013) and stage (p < 0.001). However, the risk scores were not significantly correlated to age (p = 0.102). The ability of the risk score to be used for predicting the 1- and 5-year survival rate of HCC patients is presented in Figure 7A–E . The ROC curve indicated that the risk score could be used to predict the survival of HCC patients at 1 to 3 years [areas under the curve (AUC) >0.7]. As shown in Figure 7F and G , we generated a nomogram via multivariate Cox regression analysis to predict the probability of 1-, 2-, 3-, 4-, and 5-year OS from data containing significant factors, such as age, stage, and risk score. The total points of the samples under certain conditions in the nomogram allowed an understanding of the survival situation and prediction of the 1- to 5-year survival rate of the samples. The accuracy of the prediction model was evaluated with the consistency index (C-index). The C-index was 0.718, which suggested that the accuracy was good.

Analysis of prognostic and clinical characteristics. (A) Risk score predicting the 1-year survival rate of liver cancer patients. (B) Risk score predicting the 2-year survival rate of liver cancer patients. (C) Risk score predicting the 3-year survival rate of liver cancer patients. (D) Risk score predicting the 4-year survival rate of liver cancer patients. (E) Risk score predicting the 5-year survival rate of liver cancer patients. (F) Nomogram composed of age, stage and risk score for the prediction of 1-year, 2-year, 3-year, 4-year and 5-year survival probability. (G) Calibration curve for the evaluation of the nomogram in predicting 1-year, 2-year, 3-year, 4-year and 5-year survival probability. (H) Relationship between risk score and survival time. In the survival time plot, a blue circle indicates that the sample status is alive, and the red circle indicates that the sample status is dead. (I) Heatmap presenting the expression levels of 19 m6A RNA methylation-related genes in the low- and high-risk groups. The distribution of clinicopathological features was analyzed between the low- and high-risk groups.
To explore whether these risk characteristics could function as novel prognostic risk factors, we studied the relationship among the risk score, clinicopathological characteristics (including age, stage and sex) of TCGA data samples, and survival. The results showed that only the risk score was significantly related to survival (p < 0.001) ( Table S9 and Figure 7H ). The genetic status and clinical features with significant survival in the two subgroups are shown in the heatmap ( Figure 7I ).
Analysis of differences in immune infiltration of the two subgroups
Growing evidence has demonstrated that the tumor microenvironment greatly influences the development of various tumors, such as HCC [23]. To explore the underlying mechanism of these m6A regulators in HCC, we further evaluated the abundance of different immune cells from high- and low-risk groups via the “CIBERSORT” package in R. The integrative distributions of the fractions among 22 immune cell types from the two groups are shown in Figure 8A . In the low-risk group, the immune infiltration of monocytes, NK cells, and mast cells was higher than that in the high-risk group, whereas there were more M0 macrophages in the high-risk group than in the low-risk group ( Figure 8B ).

Analysis of mutation differences between the two subgroups and correlation analysis of risk score with matrix, immune score, and mRNAsi dryness index
In addition, we performed mutation analysis. The landscape of mutation information of each gene in each sample from different groups (high- and low-groups) is exhibited in Figure 9A and B . The various colors in the waterfall plot correspond to different mutation types, and the pictures present the top 20 mutated genes in HCC with ranked percentages in the two subgroups. We found that the gene with the highest mutation frequency was TP53 (43%) in the high-risk group and TTN (24%) in the low-risk group. Moreover, we analyzed the relationship among the risk score and the stromal score, immune score and mRNAsi score via Pearson’s correlation analysis. The results revealed that there were significant differences between the risk score and the stromal score, immune score, and mRNAsi score (all p < 0.05) ( Figure 9C–E ).

Landscape of mutation profiles in HCC samples. (A) High and (B) low mutation information of each gene in each sample is shown in the waterfall plot, in which various colors with annotations at the bottom represent the different mutation types. The bar plot above the legend shows the mutation burden. Significant differences among the stromal score (C), immune score (D), mRNAsi score (E) and risk score (all p < 0.05).
Discussion
HCC is the fourth leading cause of cancer-related deaths and the sixth most common cancer worldwide. Although there has been a large advancement in the therapeutic management of HCC, patients with HCC still have poor prognosis [24]. Accumulating evidence has shown that m6A modification is linked to tumor proliferation [25], tumorigenesis, differentiation, invasion [26], and metastasis [27], and m6A modification acts both as an oncogene and an anti-oncogene in tumors. Thus, identifying m6A regulators and m6A-related genes in HCC may offer novel insight into valuable therapeutic targets.
The influence of METTL3 and METTL14 on HCC development and progression has been widely reported in previous studies [6], whereas the function of m6A-related genes is not fully elucidated. Therefore, we explored the key genes from the 1258 candidate m6A-related genes and then analyzed their correlation with the clinicopathological characteristics of HCC patients. Our study obtained 192 m6A-related candidate genes that were differentially expressed in different pathological stages of HCC patients. In addition, we found that the expression of three m6A regulators, YTHDF1, YTHDF2, and YTHDC1, was linked to the prognosis of HCC.
As RNA-binding proteins of the YTH domain family, YTHDF1 and YTHDF2 were significantly upregulated in various cancers, including HCC, which affected the cell cycle and metabolism of tumor cells. It has been reported that high YTHDF1 expression implies poor prognosis of HCC patients [28]. In addition, a previous study has found that YTHDF2 may act as a tumor suppressor to inhibit cell proliferation in HCC [16]. Recent evidence has shown that YTHDF1 may act as a prognostic factor and has an opportunity to be involved in the treatment of colon cancer patients [29]. Silencing of YTHDF2 enhances the inflammation, metastasis and angiogenesis of HCC [30]. Study found that the expression of YTHDF2 was related to infiltration of immune cells and their marker genes. In addition,YTHDF2 may have the potential effect in regulating polarization of tumor-associated macrophages, such as T-cell exhaustion, and activating T-regulatory cells [31]. YTHDC1 regulates the splicing of exons by binding to splicing factors [32]. Our study demonstrated that the expression of YTHDF1, YTHDF2, and YTHDC1 was significantly different at the four pathological stages of HCC. Therefore, the expression of m6A regulators may be strongly related to the clinicopathological features of HCC patients.
In addition, we obtained two HCC subtypes (KM1 and KM2) via consensus clustering analysis. The age, WHO classification, and survival time differed between the KM1 and KM2 subtypes (all p < 0.05) of HCC patients. In addition, we explored the differentially expressed genes between the two subgroups followed by GO function and KEGG pathway enrichment analysis. The results indicated that these genes were enriched in pathways associated with the malignant progression of HCC. However, the specific molecular mechanisms still require further study.
To explore the prognostic roles of m6A candidate genes in HCC, we used univariate analysis and LASSO regression to construct a 19-gene risk model. Among these 19 prognosis-related genes, the eight top genes were ZMYM1, BRSK2, POMT2, BBS7, TET1, THADA, GEMIN5, and DEPDC1. Previous studies have revealed that DEP domain-containing 1 (DEPDC1) is an oncogene that is upregulated and drives cell proliferation, invasion, and angiogenesis [33] but suppresses sensitivity to chemotherapy in HCC [34]. The gem nuclear organelle-associated protein 5 (GEMIN5) RNA-binding protein binds to the ribosome and is involved in global translation [35], and this alteration contributes to alternative splicing and tumor cell motility [36]. Ten–eleven translocation 1 (TET1), a widely reported DNA demethylation protein, has been reported to be expressed at low levels in HCC tissues, which facilitates HCC cell invasion by reducing the inhibitory effect of miR-494-induced HCC cell invasiveness [37]. This is consistent with the results in our study. Thus, our study provides a reference for these genes for further research.
We then divided the samples from TCGC database into high- and low-risk groups based on the median risk scores. Kaplan–Meier analysis indicated that the survival of the two subgroups was obviously distinguished. In addition, the AUCs of 1 year to 3 years among HCC patients were greater than 0.7, indicating that our risk model has the potential to predict the survival of HCC patients. We also explored the correlation among the risk score, clinicopathological characteristics (including age, stage, and sex) and survival for the samples from TCGA data. The results revealed that only the risk score was significantly related to survival, indicating the prognostic value of the risk model for HCC.
By analyzing mutation differences between high- and low-risk score groups, we found that TP53 and TTN had the highest mutation frequency in the high-risk and low-risk HCC patient groups, respectively. TP53, which has been named “the guardian of the genome,” is an important tumor-suppressor gene. The mutation in TP53 and the resultant inactivation of p53 cause the evasion of rapid tumor progression and tumor cell death [38]. In breast cancer, the mutation of TP53 was the most prevalent in patients with the basal/triple negative subtype. Jessica et al. reported that TP53 is the next most prevalent mutation, affecting 25% to 30% of HCC patients [39]. Another study has reported that TP53 pathways and regulatory miRs may be used as therapeutic targets in HCC [40]. In our study, we found that TP53 had the highest mutation frequency in the high-risk HCC patient group, which agreed with existing research and suggested the use of TP53 to assess the risk of liver cancer.
TTN, the longest-known coding gene, has been reported to be frequently mutated in some tumors, including lung adenocarcinoma and colon adenocarcinoma. It has been reported that patients with lung squamous cell carcinoma have favorable overall survival benefits from the TTN mutant type [41]. Jia et al. [42] reported that the mutation rate of TTN is 29.68% in solid tumors, and they divided tumor mutation burden into high and low groups, representing different clinical responses to immune checkpoint blockade monotherapy. However, another study has found that spontaneous mutation in the TTN gene represents a high tumor mutation burden [43]. In our study, we found that TTN had the highest mutation frequency in the low-risk HCC patient groups. Thus, we hypothesize that m6A RNA methylation regulated by TP53 and TTN may play an important role in regulating tumor proliferation and metastasis, which further influences the prognosis of HCC patients.
Conclusion
We analyzed the expression, PPI, and prognostic value of m6A-related genes in HCC. Our results indicated an close relationship between the expression of m6A-related genes and clinicopathological characteristics in HCC. We identified several m6A-related genes that may serve as novel prognostic markers in HCC patients. Our study provides integrated evidence for exploring the function of m6A in HCC and further development of therapeutics.