
Introduction
Traumatic brain injury (TBI) is a major global health concern that contributes to nearly one-third of all trauma-related deaths [1]. Extensive studies have demonstrated noticeable damage to the central nervous system evoked by secondary autodestructive insults after TBI [2–4]. Evidence of the biochemical events after secondary injury has improved the understanding of delayed cell death and brain damage [5,6]. Although some progress has been made in preventive measures and clinical care in early resuscitation, overall neurological recovery and long-term morbidity continue to pose major healthcare challenges [7].
Lysine acetyltransferases catalyze lysine acetylation, a reversible protein modification implicated in a wide variety of disease states [8,9]. The MYST-family histone acetyltransferase lysine acetyltransferase 6A (KAT6A) is involved in various fundamental cellular processes, such as gene transcription [10], cellular apoptosis [11], cardiac septum development [12], memory T-cell diversity [13] and maintenance of normal hematopoietic stem cells [14]. Abnormal regulation of KAT6A histone acetyltransferase activity or aberrant expression of KAT6A has been proposed to be associated with oncogenesis in leukemia [15] and breast cancer [16]. Moreover, a recent study has suggested a possible mechanism in which KAT6A acetylation of lysine 23 of histone H3 results in recruitment of the nuclear-receptor-binding protein TRIM24, which in turn upregulates PI3K/AKT signaling and tumorigenesis [17]. Other studies have confirmed that PI3K/AKT signaling plays an important role in secondary brain injury, through increasing neurogenesis [18], decreasing glia-mediated inflammation [19], preventing blood-brain-barrier damage [20], decreasing apoptosis [21] and inhibiting autophagy [22].
A variety of signaling pathways, such as the PI3K/AKT pathway, are involved in DNA damage and repair after TBI [22]. Activation of these signaling pathways induces a cellular-protective system that promotes cell survival after TBI. KAT6A is a critical transcription factor that regulates diverse cellular functions by modulating gene expression, and has roles in DNA damage and repair [10,14]. KAT6A also acts as a transcriptional co-activator that activates the PI3K/AKT signal pathway and further inhibits apoptosis [17]. Despite a preponderance of studies demonstrating the key roles of KAT6A in anti-apoptosis and the cell cycle, the effects of injury severity and brain temperature on the expression levels of these cell death effectors after TBI remain largely unexplored. A comprehensive understanding of the injury-induced KAT6A response, particularly the temporal profile of biochemical alterations, is crucial for designing effective therapeutic interventions.
The cytoprotective effects of hypothermia have been demonstrated by several in vivo and in vitro studies indicating that apoptosis inhibition [23] and inflammation [24] are partly due to the PI3K/AKT pathway, which involves components of the KAT6A downstream signaling pathway. However, links among the expression of KAT6A, hypothermia after injury and upstream regulation of PI3K/Akt must be elucidated. In a prior study, we identified that the expression of KAT6A is associated with survival in patients with GBM [17]. Moreover, silencing of KAT6A suppresses cell proliferation, cell migration, colony formation and tumor development in an orthotopic mouse xenograft system. Our study revealed the temporal-regional expression profiles of KAT6A after moderate and severe TBI and explored temperature-manipulation effects after TBI.
Materials and methods
Animals
Experiments were performed on male Sprague-Dawley rats (weighing from 325 g to 378 g, n = 250). All animals were kept under constant temperature (23–25°C) with 12 h light/dark cycles for 7 d before the experiments. The animals were given free access to water and food, with the exception that food was withheld overnight before surgery. The sample size was not predetermined with a statistical method, but the sample sizes were similar to those generally used in the field.
Randomization and blinding of the experimenter to treatment groups were undertaken in all animal experiments. Experimenter blinding was sufficient to control for selection bias.
Experimental groups
Individual animals were assigned to one of three groups according to surgical injury severity ( Table 1 ). The animals were subjected to standard surgical procedures except TBI in group 1 (sham). Animals in group 2, the moderate TBI group, were subjected to injury with fluid percussion (F-P), pressures of 1.8–2.2 atm. In group 3, severe TBI was induced (2.4–2.7 atm).
Experimental Groups and Outcome Measures
| Animals | RT-PCR | Western Blot |
|---|---|---|
| Sham | 4, 6, 12, 24, 48, 72 h; 1 w | 4, 6, 12, 24, 48, 72 h; 1 w |
| TBI (moderate) | ||
| Normothermia (37°C) | 4, 6, 12, 24, 48, 72 h; 1 w | 4, 6, 12, 24, 48, 72 h; 1 w |
| Hypothermia (32°C) | 12, 48 h | 24, 72 h |
| Hyperthermia (39°C) | 12, 48 h | 24, 72 h |
| TBI (severe) | ||
| Normothermia (37°C) | 4, 6, 12, 24, 48, 72 h; 1 w | 4, 6, 12, 24, 48, 72 h; 1 w |
RT-PCR, reverse transcriptase polymerase chain reaction; TBI, traumatic brain injury.
To determine the spatial and temporal expression of KAT6A mRNA and protein after moderate or severe F-P brain injury, we tested individual brain samples at various time points (4, 6, 12, 24, 48, 72 h and 7 d) after moderate F-P injury or severe TBI (n = 5 animals per group).
For the next phase, we investigated the systemic effects of post-traumatic brain temperature on KAT6A mRNA and protein expression levels. Animals were treated with different temperatures (32°C, 37°C and 39°C) for 4 h immediately after TBI, as well as at 39°C after sham injury, with cooled air or a heating lamp. All animals were sacrificed at 12 or 48 h after TBI for analysis of relative mRNA expression or assessment of protein expression at 24 and 72 h after TBI.
Surgical preparation
Rats were anesthetized with 2% halothane with nitrous oxide/oxygen mixture (70%/30%), and this was followed by tracheal intubation and positive-pressure ventilation. Blood pressure and blood gas were continually monitored with PE50 polyethylene tubing cannulated in the femoral artery and vein on the right side. Animals were sutured after cannulation, then turned into the prone position. The sagittal scalp was incised. A 4.8-mm-diameter opening was made 2 mm to the left side of the midline between the bregma and lambda. For fixation, two screws were placed at 1 mm separately, rostral to the bregma and caudal to the lambda. A Luer Lok needle hub (Becton Dickinson, Mountain View, CA) was placed into the skull hole and cemented in place with superglue and dental acrylic. A 1.5-mm burr hole was then drilled at the middle of the occipital bone. The brain temperature probe, with the assistance of a 1.5-inch needle guide, was adhered to the surface of the skull over the burr hole with cyanoacrylate adhesive and dental acrylic. The mean arterial blood pressure (MABP) was recorded continuously during the entire experiment. Blood gases were measured before injury and within 15 min after injury.
Fluid percussion TBI
A customized fluid percussion device was used to produce TBI, as previously described [25]. Briefly, injury was induced by descending of a metal pendulum striking the piston, injection of a small volume of saline epidurally into the closed cranial cavity, and production of brief displacement and deformation of the neuronal tissue. The magnitude of the pressure pulse was measured by a pressure transducer, stored on an oscilloscope, and later converted to atmospheres with an extracranial transducer (Statham PA 85-100; Gould, Oxnard, CA) and further recorded on a storage oscilloscope (Tektronix 5111; Tektronix, Beaverton, OR). Animals were injured with a moderate (1.8–2.2 atm) or severe (2.4–2.7 atm) magnitude of injury.
Temperature manipulation
A digital electronic thermometer was used to monitor the temperature of the frontal cortex of the brain (model DP 80/HYP-033-1-T-G-60-SMP-M; Omega Engineering, Inc., Stamford, CT). Briefly, the temperature probe was placed at 4.0 mm to the surface of the skull ventrally. The probe was removed before fluid percussion injury and replaced immediately afterward. Rectal temperatures were measured with an electronic thermometer with an analog display (model 43 TE/400; YSI, Inc., Yellow Springs, OH) was used to measure rectal temperature. The initial brain temperature remained at 37°C before the hypothermic treatment. Hypothermia was induced as previously described [26]. In brief, brain temperatures were cooled to 32°C by immersion of the body of the animal in an ice-cold water bath. The animals were placed in plastic bags (with head exposed) in advance to protect the skin and fur from direct contact with water. The temperature was controlled by removing animals from the water bath as long as the brain temperature had decreased to within the range of 2°C of the target. The target temperature was reached in approximately 15 min. The target temperature was maintained for 4 h under general anesthesia at room temperature through intermittent application of ice packs as necessary. Gradual rewarming to normothermic levels within 90 min according to the reported procedures was used to avoid rapid rewarming and influencing secondary injury processes [27]. A heating blanket was used for as long as 4 h to achieve brain temperatures of 37°C and 39°C under general anesthesia.
Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) was performed to explore the time course of mRNA for KAT6A at multiple time points (4, 6, 12 h, 1–3 d and 1 week) after injury. Total RNA was isolated from the tissue harvested from the hippocampus with TRIzol reagent (Invitrogen, Carlsbad, CA, USA). qRT–PCR with a Lightcycler® System (Roche Diagnostics Corporation) was used to measure mRNA levels of KAT6A. A total of 2 μg of RNA was used in the following procedures, as previously described. The following primer pairs were used: rat KAT6A, forward: 5′-GAGGCCAATGCCAAGATTAGAAC-3′ and 5′-AGCTTGACTAAAGGGCTGTC-3′. Separate PCR reactions were conducted for each transcript and comprised cDNA and SYBR Premix Ex Taq (TaKaRa, Ltd., Japan). Relative gene expression was determined on the basis of the ΔCt values between the gene of interest and the housekeeping gene GAPDH, compared with the mean ΔCt values for the PBS buffer control group.
Western blotting
KAT6A-3 protein (220 kD) from individual animals was measured with western blotting at the indicated time points after TBI. Briefly, total protein concentration was determined with a BCA protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA). Rabbit anti-KAT6A antibody (1:100, Abnova, PAB8745) and mouse anti-human β-actin (1:5 k, Abcam, ab8226, Cambridge, MA, USA) were incubated overnight at 4°C. After rinsing, samples were labeled with the secondary antibody IRDye 800CW goat anti-rabbit (1:2 k, LI-COR Biosciences, 2651127, Lincoln, Nebraska USA) and IRDye 800CW goat anti-mouse antibody (1:5 k, LI-COR Biosciences, 2782997, Lincoln, Nebraska USA). KAT6A protein was detected with a LI-COR imaging system (LI-COR Biosciences, Lincoln, Nebraska USA) and exposure to Kodak diagnostic film (Eastman Kodak Co., Rochester, NY, USA). Gels were imaged with an Agfa DuoScan instrument (Agfa Corp., Ridgefield Park, NJ, USA) and subsequently quantified with Alpha Ease (Alpha Innotech Corporation, San Leandro, CA, USA). Samples from the independent hemispheres from all animals at each time point were run on gels simultaneously to allow for comparisons between groups. The relative protein expression was quantified by normalization to β-actin.
Immunofluorescence and imaging
Immunofluorescence was performed on free-floating tissue sections. Samples were permeabilized for 1 h in 0.1% Triton X-100 (PBS) buffer to avoid non-specific labelling. Dual labeling was achieved via the simultaneous incubation of primary antibodies at 4°C for 24 h. Anti-KAT6A (1:25, Abnova, PAB8745), anti-NeuN (1:1 k, Chemicon, ABN91) and anti-GFAP (1:1 k, Chemicon, SAB2107063) were used. After rinsing, samples were treated with Cy3- and Cy2-conjugated anti-mouse IgG (1:500) from Jackson ImmunoResearch Labs. Samples treated with only secondary antibodies served as negative controls. Sections mounted in Mowiol medium 4-88 (Calbiochem Corp., La Jolla, CA, USA) were imaged with a Bio-Rad MRC 1024 laser scanning confocal microscope (40× oil immersion lens). The obtained images were processed in Adobe Photoshop 9.0 (Adobe Systems, San Jose, CA).
Statistical analysis
Physiological variables between groups were compared with one-way ANOVA. Statistics on KAT6A data were performed with two-way ANOVA. Tukey tests were used for post hoc comparisons. Statistical significance was indicated by p < 0.05. Data are presented as mean ± SEM. Normality was verified with Shapiro-Wilk tests, and homogeneity of variance was verified with Brown-Forsy the tests in multiple groups.
Results
All physiological variables except temperature were within normal ranges across groups (TBI normothermic, TBI hyperthermic TBI hypothermic and sham groups; Table 2 ).
Physiological variables
| Values of Physiological Variables | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Groups | Sham | Norm 6 h | Hypo 6 h | Norm 12 h | Hypo 12 h | Norm 24 h | Hypo 24 | Norm 72 h | Hypo 72 h |
| Pre-trauma 15 min | |||||||||
| PH | 7.44 ± 0.12 | 7.43 ± 0.8 | 7.44 ± 0.06 | 7.44 ± 0.07 | 7.44 ± 0.11 | 7.43 ± 0.05 | 7.45 ± 0.05 | 7.45 ± 0.04 | 7.44 ± 0.07 |
| PCO2 (mm Hg) | 38.65 ± 1.57 | 38.79 ± 1.47 | 38.49 ± 1.62 | 37.78 ± 1.29 | 38.69 ± 1.48 | 38.26 ± 0.99 | 39.09 ± 1.58 | 38.44 ± 1.37 | 38.21 ± 1.80 |
| PO2 (mm Hg) | 137.77 ± 4.87 | 136.82 ± 3.64 | 135.73 ± 2.29 | 138.87 ± 5.83 | 138.79 ± 6.77 | 138.54 ± 6.088 | 136.39 ± 4.56 | 137.21 ± 5.29 | 139.03 ± 4.37 |
| MABP (mm Hg) | 115.44 ± 3.98 | 116.77 ± 4.29 | 118.82 ± 3.71 | 18.29 ± 3.84 | 118.97 ± 3.55 | 1119.69 ± 4.98 | 116.91 ± 4.57 | 116.43 ± 4.01 | 119.83 ± 4.39 |
| Brain t (°C) | 36.88 ± 0.11 | 36.80 ± 0.13 | 36.81 ± 0.07 | 36.69 ± 0.16 | 36.77 ± 0.08 | 36.82 ± 0.12 | 36.80 ± 0.17 | 36.82 ± 0.14 | 36.80 ± 0.09 |
| Rectal t (°C) | 36.77 ± 0.11 | 36.79 ± 0.19 | 36.89 ± 0.15 | 36.80 ± 0.15 | 36.83 ± 0.18 | 36.88 ± 0.09 | 36.79 ± 0.18 | 36.79 ± 0.18 | 36.82 ± 0.17 |
| Post-trauma 15 min | |||||||||
| PH | 7.43 ± 0.06 | 7.42 ± 0.07 | 7.43 ± 0.10 | 7.46 ± 0.08 | 7.46 ± 0.06 | 7.43 ± 0.11 | 7.44 ± 0.10 | 7.44 ± 0.08 | 7.46 ± 0.07 |
| PCO2 (mm Hg) | 38.22 ± 1.61 | 38.88 ± 1.76 | 38.59 ± 1.89 | 38.71 ± 1.21 | 38.49 ± 1.86 | 39.51 ± 1.57 | 38.39 ± 1.49 | 38.41 ± 1.71 | 38.91 ± 1.88 |
| PO2 (mm Hg) | 137.66 ± 5.29 | 137.98 ± 4.69 | 139.65 ± 5.21 | 139.33 ± 4.55 | 137.81 ± 6.39 | 137.99 ± 5.24 | 137.81 ± 3.65 | 139.28 ± 6.74 | 137.89 ± 7.01 |
| MABP (mm Hg) | 117.88 ± 4.21 | 119.71 ± 3.78 | 120.01 ± 3.36 | 119.91 ± 3.48 | 119.76 ± 3.97 | 118.39 ± 3.35 | 118.29 ± 4.11 | 118.04 ± 4.18 | 120.69 ± 4.21 |
| Brain t (°C) | 36.71 ± 0.11 | 36.77 ± 0.16 | 32.21 ± 0.44﹡ | 36.75 ± 0.09 | 32.43 ± 0.71﹡ | 36.81 ± 0.14 | 32.46 ± 0.33﹡ | 36.77 ± 0.19 | 32.49 ± 0.51﹡ |
| Rectal t (°C) | 36.78 ± 0.11 | 36.99 ± 0.08 | 33.66 ± 0.51﹡ | 36.87 ± 0.09 | 33.31 ± 0.37﹡ | 36.93 ± 0.13 | 33.49 ± 0.29﹡ | 36.78 ± 0.11 | 33.83 ± 0.62﹡ |
Data are presented as mean ± SEM. Blood pH, blood gases (pCO2 and pO2, mm Hg), mean arterial blood pressure (MABP, mm Hg), brain temperature and rectal temperature (brain t and rectal t °C) were monitored 15 min before trauma (pre-trauma) and 15 min post-trauma. All physiological variables, with the exception of temperature, were within normal ranges for all TBI normothermic groups and TBI hypothermic groups. Bold values indicate p < 0.001. *Indicates significant hypothermia.
Downregulation of KAT6A mRNA after moderate TBI
The KAT6A mRNA expression was quantified with qRT-PCR in the ipsilateral and contralateral hemispheres in a time course after TBI ( Figure 1 ). In the sham group, the KAT6A mRNA remained at a steady level within the examined duration ( Figure 1 ). At 4 h after TBI, the KAT6A mRNA levels (in the ipsilateral and contralateral cortex or hippocampus) in the severe TBI group were markedly lower than those in the sham group (p < 0.01). We also observed a further decrease in the mRNA level after TBI, which gradually reached a minimum 2 d after TBI. The results showed that the mRNA level of KAT6A in the ipsilateral side approximately 12 h after TBI was significantly decreased. Similarly, in the moderate TBI group, the KAT6A mRNA expression was significantly lower than that in the sham group (4 h after TBI) (p < 0.01). A similar pattern was found in the severe TBI group, in which with the minimum value was synchronously reached in the cortex and hippocampus on both sides. Interestingly the decrease was significantly attenuated with respect to that in the severe TBI group. The minimum mRNA expression levels of KAT6A significantly decreased at the injured ipsilateral site and uninjured (contralateral) hemispheres (both cortex and hippocampus; severe TBI: 75.10 ± 3.10%, 79.69 ± 3.30%; moderate TBI: 89.69 ± 2.40%). KAT6A mRNA expression in the injured hemisphere was higher in the moderate TBI group than the sham group at all time points within 7 d (p < 0.01). However, 1 week after TBI, the KAT6A mRNA level of all groups decreased and showed no significant differences across the regions (0.011 ± 0.001). In the sham control group, the mRNA levels of KAT6A stayed at a low level in ipsilateral cortex (0.011 ± 0.001) and hippocampus (0.011 ± 0.002) as well as contralateral cortex (0.009 ± 0.001) and hippocampus (0.012 ± 0.001) (p > 0.05).
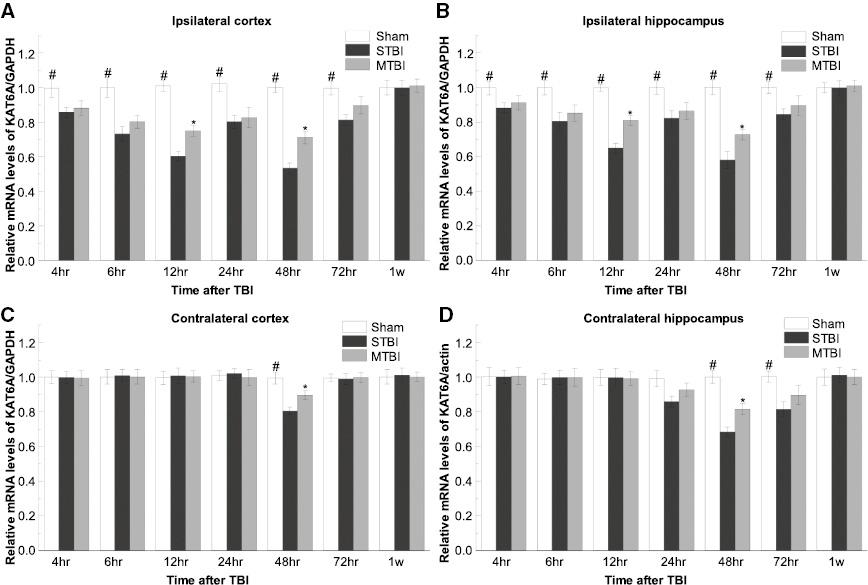
Spatial and temporal expression of KAT6A mRNA after moderate TBI. Comparison of relative levels of KAT6A mRNA at ipsilateral (A and B) and contralateral (B and D) hemispheres from the sham-operated, severe and moderate TBI groups (#p < 0.01 for STBI and MTBI groups versus SHAM; *p < 0.01 for MTBI group versus STBI, n = 5 per group). SHAM, sham injured; STBI, severe traumatic brain injury; MTBI, moderate traumatic brain injury.
Our results suggested that TBI induces the downregulation of KAT6A mRNA expression, and injury severity determines the range of the decline of KAT6A mRNA levels after TBI.
Decreased expression of KAT6A proteins after moderate TBI
We then examined the expression of KAT6A at the protein level via western blotting ( Figure 2 ), and further investigated the expression and localization of KAT6A after TBI by immunohistochemical staining. KAT6A expression in sham-treated animals remained at a relatively constant low level in the ipsilateral cortex (0.012 ± 0.001) and hippocampus (0.009 ± 0.001), contralateral cortex (0.011 ± 0.001) and hippocampus (0.012 ± 0.002) (p > 0.05). This finding was consistent with our immunofluorescence results ( Figures 3 and 4 ). KAT6A co-localized with both glial fibrillary acidic protein (GFAP) and neuronal nuclei-specific antibody (NeuN) in the hippocampus.
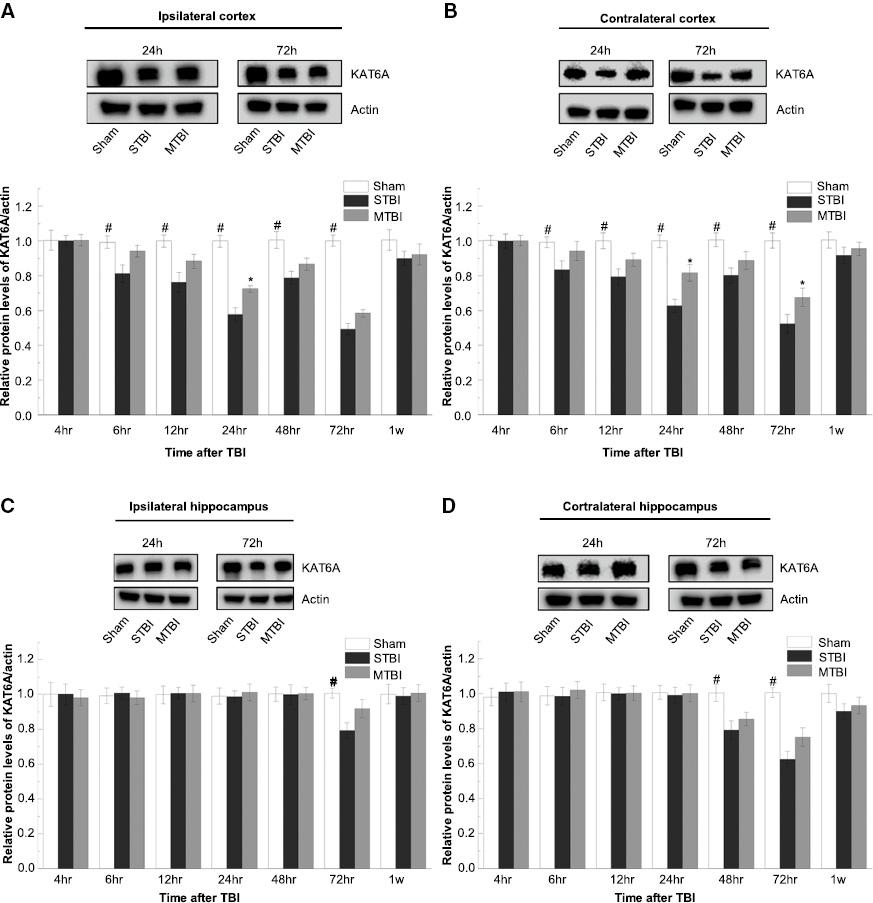
Temporal and regional expression of KAT6A proteins after MTBI. Western blots of KAT6A in the ipsilateral (A and C) and contralateral (B and D) hemispheres after TBI are shown. The levels of protein expression were calculated by normalization to β-actin (#p < 0.01 for STBI and MTBI groups versus SHAM; *p < 0.01 for MTBI group versus STBI, n = 5 per group).
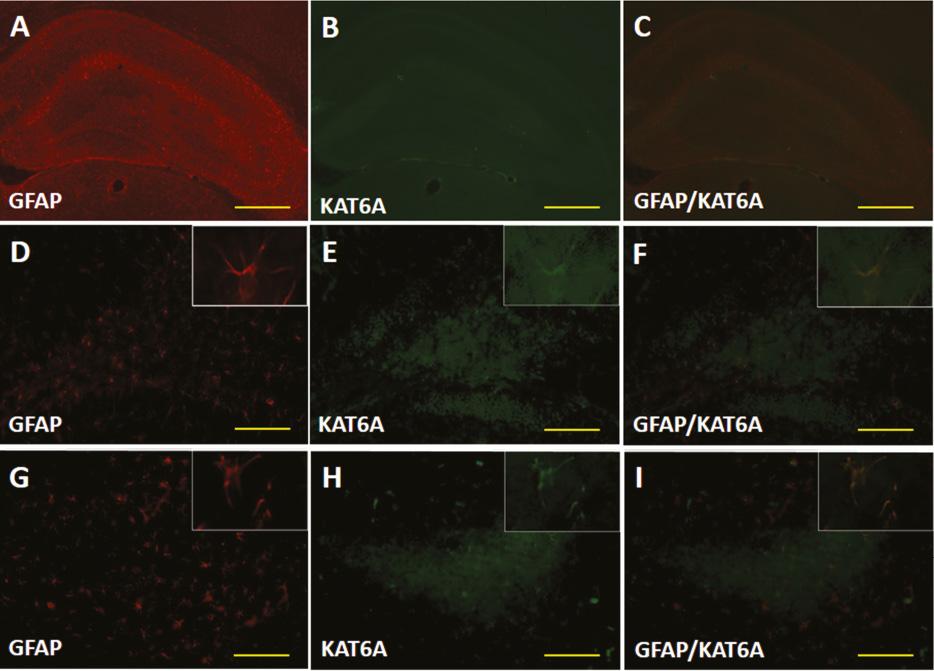
(A–I) Double fluorescence images of KAT6A, and cells identified with a specific marker (GFAP). KAT6A co-localized with GFAP in the hippocampus after TBI (scale bar = 30 mm).
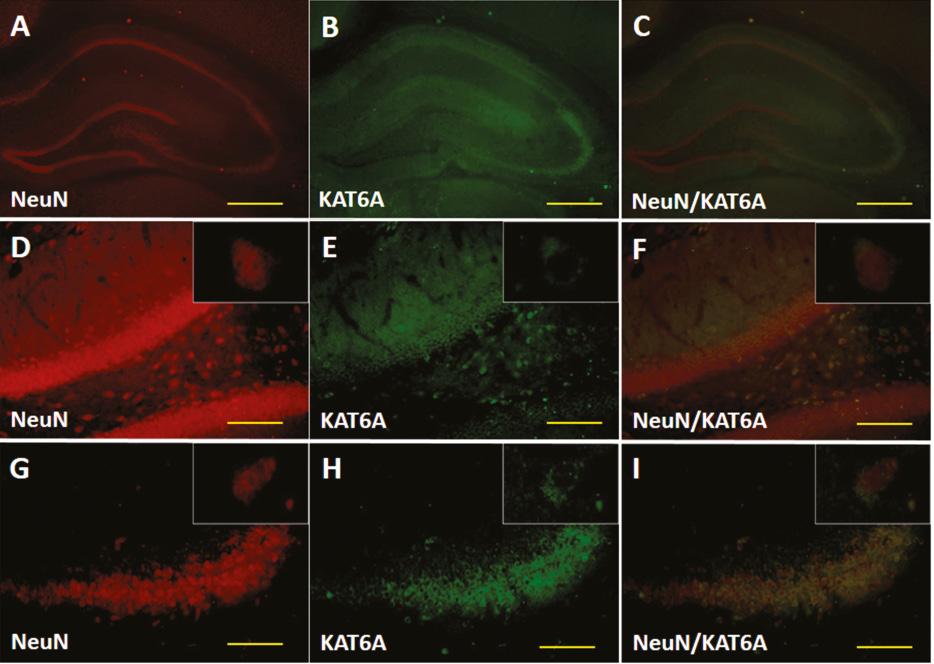
(A–I) Double fluorescence images of KAT6A, and cells identified with a specific marker (NeuN). KAT6A co-localized with NeuN in the hippocampus after TBI (scale bar = 30 mm).
At 6 h after TBI, the amounts of KAT6A in both the severe and moderate TBI groups were significantly lower than those in the sham group (p < 0.01). KAT6A protein in the ipsilateral and contralateral hemispheres of the severe TBI and moderate TBI groups continued to decrease to a minimum at approximately 72 h after TBI ( Figure 2 ). In line with the qRT-PCR results, the second temporal minimum of KAT6A protein expression was found in the ipsilateral side at 24 h after TBI. For the cortex and hippocampus in the injured (ipsilateral) hemisphere, interestingly, the decrease in the KAT6A protein level in the moderate TBI group was smaller than that in the severe TBI group (p < 0.01). The minimum protein levels of KAT6A in injured (ipsilateral) and uninjured (contralateral) hemispheres (both cortex and hippocampus) decreased to 84.27 ± 2.60% and 77.36 ± 3.40% in the severe TBI group, and 86.38 ± 2.70% and 83.11 ± 3.80% in the moderate TBI group, respectively.
Distinct KAT6A mRNA expression changes after moderate TBI followed by post-traumatic temperature manipulation
The levels of KAT6A mRNA in the normothermic and hyperthermic groups were significantly lower than those in the hypothermic group at 12 h after TBI (p < 0.01), whereas no significant difference was found between the hyperthermic and normothermic groups (p > 0.05).
However, the level of KAT6A mRNA in the hyperthermic group was significantly lower than that in the normothermic group at 48 h after TBI; in contrast, post-traumatic hypothermia, in comparison to hyperthermia, significantly upregulated the mRNA levels in the ipsilateral and contralateral hemispheres.
In contrast, in the sham-injured group, KAT6A mRNA levels were maintained at a constant low value (p > 0.05). Temperature manipulation did not significantly affect the KAT6A mRNA levels at 12 h and 48 h ( Figure 5 ).

KAT6A mRNA expression at 12 h and 48 h after MTBI followed by 4 h of post-traumatic temperature manipulation. Normothermia (37°C), hyperthermia (39°C) and hypothermia (32°C) significantly affected KAT6A mRNA expression in the ipsilateral (A and B) and contralateral (C and D) hemispheres, compared with these same regions at 48 h. An additional hyperthermic group (39°C) of sham-injured animals was included to demonstrate that the upregulation of KAT6A in hyperthermic post-traumatic rats was due to the combination of TBI and temperature rather than temperature alone (#p < 0.01 for the hyperthermic TBI and normothermic TBI groups versus the hypothermic TBI group; *p < 0.01 for the hypothermic TBI group versus the normothermic TBI group, n = 5 per group).
KAT6A protein expression under hypothermia and hyperthermia after moderate TBI
The KAT6A protein levels in the normothermic and hyperthermic groups at 24 h after TBI were significantly lower than those in the hypothermic group (p < 0.01). However, no significant difference was observed in protein levels between the hyperthermic and the normothermic groups (p > 0.05).
Interestingly, the KAT6A protein expression in the hypothermic group at 72 h was significantly higher than that in the normothermic and hyperthermic groups. In contrast, post-traumatic hyperthermia, as compared with normothermia, significantly decreased the KAT6A expression.
In the sham-injured group, in line with the KAT6A mRNA results, the protein levels were maintained at a steady low level (p > 0.05). The altered temperature had limited influence on KAT6A protein expression at 24 h and 72 h ( Figure 6 ).

KAT6A protein expression at 24 h and 72 h after MTBI followed by 4 h of post-traumatic temperature manipulation. Temperature control, such as normothermic (37°C) treatment, hyperthermic (39°C) treatment and hypothermic (32°C) treatment, influenced KAT6A protein expression in the ipsilateral (A and B) and contralateral (C and D) hemispheres with respect to comparable regions at 72 h. An additional hyperthermic group (39°C) of sham-injured animals was also included to demonstrate that the enhanced expression of KAT6A in hyperthermic post-traumatic rats was caused by the combination of TBI and temperature rather than temperature alone (#p < 0.01 for the hyperthermic TBI and normothermic TBI groups versus the hypothermic TBI group; *p < 0.01 for the hypothermic TBI versus the normothermic TBI group).
Discussion
Our data indicated that moderate TBI, compared with sham treatment, triggered significantly lower KAT6A mRNA and protein expression in the cortex and hippocampus within 1 week (p < 0.01). Furthermore, considerably reduced the KAT6A mRNA/protein expression levels in the same regions of the ipsilateral hemisphere were detected in the severe TBI group, compared with the sham group (p < 0.01). However, before the downregulated expression at 48 h (mRNA) and 72 h (protein), a significant upregulation was observed at 12 h (mRNA) and 24 h (protein), with respect to the levels in the moderate TBI group. Moreover, post-traumatic hypothermia, as compared with normothermia and hyperthermia, significantly inhibited the downregulation of KAT6A mRNA levels at 12 h and 48 h in the cortex and hippocampus in the ipsilateral hemisphere. However, significantly lower KAT6A protein levels than those the normothermic group were observed at 24 h and 72 h in these same brain sections. Post-traumatic hyperthermia, as compared with normothermia, resulted in a significant decrease in KAT6A at both the mRNA and protein levels (at 48 and 72 h) in the cortex and hippocampus. This biphasic regulation of KAT6A in the post-TBI hyperthermic group may be associated with the combined effects of both TBI and temperature.
Histone acetylation is generally believed to be essential for DNA repair through promoting the activity of DNA replication origins through the recruitment of histone acetyltransferases [28]. Histone lysine N-acetylation is catalyzed by histone acetyltransferases, which are more generally termed lysine acetyltransferases [29]. The lysine acetyltransferase nomenclature, although widely used, has not become generally accepted. KAT6A binds and acetylates histones via its MYST domain [30]. Aberrant expression of KAT6A is correlated with oncogenic activity in acute myeloid leukemia (AML). KAT6A is bound with the CREB binding protein (CBP), leading to nuclear translocation [15]. The KAT6A nucleosome-binding motif and TIF2-mediated recruitment of CBP may play an important role in gene translocation [23,31]. We found that moderate TBI significantly decreased KAT6A expression at both the mRNA and protein levels. Therefore, KAT6A may be important in DNA repair after TBI.
Kindle et al. first showed that KAT6A is a strong inhibitor of the transcriptional activities of CBP-dependent activators, such as nuclear receptors and p53 in AML [32]. After DNA damage, p53 induces either cell-cycle arrest or apoptosis [13]. Rokudai then showed that KAT6A forms a complex with p53, thereby inducing p21 expression and cell-cycle arrest [13]. The suggested underlying mechanism is that KAT6A acetylates lysine 23 of histone H3, thus leading to recruitment of the nuclear-receptor-binding protein TRIM24, which in turn upregulates PI3K/AKT signaling and tumorigenesis [17]. Inhibition of p53-p21 signaling pathways promotes PI3K/AKT, which is also part of the crucial mechanism of secondary brain injury after TBI. Extensive studies in the past 20 years have shown that inhibiting p53-p21 signaling decreases apoptosis [33], and improves histological and functional outcomes [34] in secondary brain injury. The PI3K/AKT signaling pathway in secondary brain injury has been confirmed to have roles in promoting neurogenesis [18], decreasing glia-mediated inflammation [19], protecting against blood-brain-barrier damage [20], preventing apoptosis [21] and inhibiting autophagy [22]. However, studies examining the mechanisms through which KAT6A mediates the PI3K/AKT signaling pathway in TBI are lacking. Thus, further studies are required to verify the role of KAT6A in TBI.
We next examined the relationship between KAT6A and post-TBI hypothermia. Our data demonstrated that, under similar injury severity, KAT6A protein and mRNA expression profiles differed between hemispheres and even within various regions of the same hemisphere. In the moderate TBI group and severe TBI group, two temporal minima in the expression of KAT6A mRNA and protein levels were found in the ipsilateral hemisphere (cortex and hippocampus). Although the first minimum was larger than the second, a single minimum in KAT6A mRNA and protein levels was found in the contralateral hemisphere. The first minimum levels of KAT6A mRNA and protein in the ipsilateral cortex were lower than those in the ipsilateral hippocampus. This difference might corelate with the increase seen in the early period (mRNA and protein expression at 12 h and 24 h, respectively) in the ipsilateral cortex. Given direct mechanical damage, the separation between the ipsilateral hippocampus and the damaged site (the ipsilateral cortex) was likely to have attenuated the increase. This result was consistent with previously reported findings [35]. Regarding the second KAT6A mRNA and protein minimum values in the ipsilateral cortex and hemisphere (observed at 48 h and 72 h for mRNA and protein, respectively), the decreased KAT6A expression negatively correlated with the degree of injury. The lower KAT6A mRNA and protein levels in the contralateral hippocampus than the contralateral cortex might have been due to secondary damage (p < 0.05 for mRNA at 48 h and for protein at 72 h), given that the hippocampus is more sensitive to secondary damage, as has been reported after ischemia [36,37]. Therefore, our findings suggest that the first peak might have been a result of direct damage, whereas the higher value of the second minimum might have largely been due to secondary injury from autodestructive insults, e.g., ischemia.
Because TBI is a highly complex injury to the brain with multiple consequences, identifying the pathophysiological mechanisms is challenging but important to improve outcomes in clinical settings [2]. The pathophysiology of TBI is multidimensional and involves many injury mechanisms. Each mechanism may markedly affect neuronal dysfunction or cell death. This complexity is a major factor in determining whether promising treatments can be identified and translated to clinical settings. Pioneering studies on moderate hypothermia after TBI as a neuroprotective strategy in rodents have been performed by Dietrich and colleagues [38]. Cognitive improvements after TBI with hypothermia have been demonstrated [39]. Subsequently, further evidence indicated the efficacy of application of hypothermia; however, controversy persists regarding the effects on human patients [40]. Kuo and colleagues have shown that brain cooling without interference with the core temperature in rats significantly attenuates TBI-induced cerebral infarction and motor deficits, and subsequently significantly promotes angiogenesis and neurogenesis [41]. Our previous study has shown that programmed cell death (apoptosis and autophagy) signaling cascades are affected by hypothermia in a rat TBI model. Similar findings have been reported by others [42]. Beyond the established benefits in neuronal survival and axonal injury, hypothermia decreases breakdown of the blood-brain barrier and increases oligodendrocyte survival [43,44]. Thus, mild hypothermia appears to activate neurogenesis, decrease inflammation, protect against blood-brain-barrier damage, prevent apoptosis and inhibit autophagy via KAT6A, through the inhibition of p53-p21 and the promotion of PI3K/AKT signaling pathways. Our findings strongly suggest that injury severity and temperature are important factors regulating KAT6A expression after TBI. The protection provided by hypothermia may be at least partially due to KAT6A upregulation.
In summary, we found that KAT6A mRNA and protein expression levels varied between hemispheres and within distinct regions from the same hemisphere. The early expression of KAT6A is positively correlated with injury, whereas the negative correlation in later stages after injury indicates the complexity of secondary brain injury in TBI. The influence of mild hypothermia on the expression of the protective protein KAT6A after TBI further demonstrated the effectiveness of this protein’s complex role in secondary brain injury after TBI. Further investigations on the underlying mechanisms should therefore be performed. We propose that modest hypothermia may be an effective therapeutic method, particularly in early phases post-TBI, to moderate secondary brain injury via regulating the expression of KAT6A.