
INTRODUCTION
Influenza viruses cause respiratory infections in humans that range from asymptomatic to deadly disease. Regular epidemics and pandemics make the influenza virus a global health threat. The nucleoprotein (NP) is a multifunctional viral protein with important structural and functional roles [1, 2]. It plays a critical role in viral replication. NP is involved in important functions such as RNA packaging [3], nuclear trafficking, transcription, and replication of vRNA [4, 5]. Each genomic segment of the influenza virus vRNA is associated with multiple NP molecules and a single polymerase complex (with PB2, PB1, and PA proteins) forming a viral ribonucleoprotein (vRNP) complex. The NP binds to single-stranded vRNA in a non-specific manner [6] with a periodicity of one NP molecule per every 24 bases [7, 8]. The NP contains the nuclear localization signal (NLS), which is essential for the import of vRNP complex to the nucleus where the mRNAs are transcribed. The newly translated NPs participate in the protection of genomic RNA from degradation. The structure of NP is relatively conserved (less than 11% variance) among the different influenza A strains.
Therefore, NP is an appealing target for the development of antiviral drugs [9] and an attractive candidate as a component for broad-spectrum influenza vaccines [10, 11]. For example, it was shown that immunization with a plasmid DNA expressing influenza NP followed by boosting with adenovirus expressing NP leads to the induction of the protective immunity against challenge with highly pathogenic H5N1 influenza virus in animal models [12].
In addition, the NP serves as a target antigen for the diagnosis of influenza virus infections [13, 14].
Therefore, the development of efficient methods for recombinant NP production is an important goal. The expression of recombinant proteins in bacteria and/or eukaryotic cells is the subject of intensive studies [15, 16]. The most prevalent approach for the acceleration of the recombinant protein expression is codon optimization [17]. The successful codon-optimized expression of NP in E. coli and in Sf9 insect cells was shown by Huang et al. [18] and Yoon et al. [19], respectively. At the same time, as it was noted in a number of publications [16, 20, 21], codon optimization can affect the co-translational protein folding, protein configuration and function. Several recent studies proved that the use of the messenger RNA containing rear codons decelerates translation and at the same time ensures the proper co-translational protein folding and consequently post-translational modification and function of expressed proteins [15, 21-23]. Codon optimization can lead to disruption of the co-translational protein folding and consequently to the formation of proteins with impaired functions or non-functional proteins. Konczal et al. [24] showed that introduction of rare codons back to the codon-optimized DNA sequence leads to overall slower translation, but at the same time to enhanced recovery of the soluble protein since it provides the proper co-translational folding of the recombinant protein. Codon optimization in this case led to enhancement of the total protein yield but to lower yield of the desired soluble protein. Therefore, the efficient expression of recombinant proteins using native DNA remains an important goal.
In this study, we showed that influenza A and B virus NP genes could be efficiently expressed in E. coli without resorting to codon-optimization. Both NPs produced by this method were monomers, were fairly well soluble, and possessed specific immune reactivity.
MATERIALS AND METHODS
Selection of influenza strains and the production of purified viral concentrates
The influenza A/Brisbane/10/2007 (H3N2) and B/Brisbane/46/2015 viruses were used for the production of recombinant NPs. These strains were obtained from the Smorodintsev Research Institute of Influenza (St. Petersburg, Russia) collection. The influenza A and B viruses were grown in the allantoic cavity of 10-day old embryonated chicken eggs at 37°C and 34°C, respectively. Allantoic fluid was harvested in 48 h for influenza A and in 72 h for influenza B strain post inoculation. Virus-containing allantoic fluids were concentrated and the viruses were purified by ultracentrifugation (Beckman, Type 19 rotor) using sucrose density gradients [25] and stored at -80°C.
Construction of pET22b+NP A and pET22b+NP B expression plasmids
Total RNA was isolated from the corresponding purified viral concentrates using the Rneasy Plus Mini Kit (Qiagen, UK). The first strand of cDNA was synthesized using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, USA) with random primers. The cDNA corresponding to both NP genes was amplified using forward primers with a NdeI restriction site and the reverse primers containing the XhoI restriction site (Table 1).
| Primer name | Sequence (5’ → 3’) |
|---|---|
| NPA F NdeI | CATATGGCGTCCCAAGGCACCAAACG |
| NPA R XhoI | CTCGAGATTGTCGTACTCTTCTGCATTGTCTC |
| NPB F NdeI | CATATGTCCAACATGGATATCGA |
| NPB R XhoI | CTCGAGATAATCGAGGTCATCAT |
Polymerase chain reaction (PCR) was conducted with Encyclo polymerase (Evrogen, Russia) in Bio-Rad C1000 machine (USA). PCR for the influenza B NP gene was challenging due to the fact that the reverse primer had homology with a short section in the middle of the nucleotide sequence of this gene (from 299 to 306 bp). That led to the generation of a predominantly shorter product of 306 bp instead of the desired product of 1682 bp under a variety of conditions. This trend persisted despite the use of different quantities of template, extension temperature gradients, and usage of different polymerases. Therefore, the reamplification of cDNA was not used. Cloning of the influenza A NP gene was straightforward.
The NP A and NP B PCR products were inserted into the pET22b+ commercial vector (Novagen) (Fig. 1). PCR products were gel-purified using the Cleanup Standard Kit (Evrogen, Russia) and digested by two restriction enzymes NdeI and XhoI (Thermo Fisher Scientific, USA). The pET22b+ vector was also digested by the same enzymes, followed by the ligation of the vector/insert mixture.
Electrocompetent E. coli BL21(DE3) cells were transformed by the ligation mix, and colonies were selected on LB/ampicillin (100 μg/ml) plates. After the confirmation that bacteria carry the expression plasmid by PCR, the corresponding colonies were selected. The sequencing of the pET22b+ NP A and pET22b+ NP B constructs was performed using ABI PRISM 3100 Genetic Analyzer and their identity was confirmed by comparison to the original sequences of influenza A/Brisbane/10/2007 virus NP (PubMed accession number KJ609215.1) and influenza B/Brisbane/46/2015 NP (GISAID accession number EPI772456) genes.
Expression and purification of the fusion proteins
Selected clones were cultured overnight on LB/ampicillin plates at 37°C. The obtained cultures were dissolved in the pre-warmed fresh LB with ampicillin (100 μg/ml) to form a solution with OD600 of 0.1 and then grown in flasks at 37°C with shaking until the OD600 of the solution reached 0.9.
Expression of the NP A and the NP B was initiated by the addition of IPTG to a final concentration of 0.1 mM. The cultures were grown for 3 h at 37°C with 250 rpm shaking. Bacterial pellets were obtained from cultures by centrifugation at 5000 g (Eppendorf, 5804R) for 15 min. Cell pellets were resuspended in 40 ml of lysis buffer (50 mM sodium phosphate, 500 mM sodium chloride, 20 mM imidazole) and 1 mM of phenylmethylsulfonyl fluoride (PMSF, pH 8.0) protease inhibitor. The cells were subsequently lysed by sonication on ice and centrifuged at 13,000 g for 30 min to remove cell debris and insoluble proteins.
Supernatants containing soluble fractions of recombinant NPs (recNPs) were filtered through 0.45 μm PES syringe filters. A HisTrap HP one milliliter columns (GE Healthcare, USA) were equilibrated with 5 ml of binding buffer (50 mM sodium phosphate, 500 mM sodium chloride, and 20 mM imidazole, pH 8.0) at a flow rate of 1 ml/min. Filtered supernatants were loaded on the columns at the same flow rate. Then columns were washed with 20 ml of binding buffer in order to remove unbound material followed by elution with 10 ml of elution buffer (50 mM sodium phosphate, 500 mM sodium chloride, and 500 mM imidazole, pH 7.8) at the same flow rate. Protein-containing fractions were collected, combined, and passed through a ten milliliter BioScale Mini P-6 desalting cartridge (Bio-Rad, USA), preliminary equilibrated with PBS buffer, at a flow rate of 5 ml/min. Protein concentrations were determined by Lowry assay, and the purity of the recNPs were determined by SDS-PAGE and densitometry.
HPLC purification of recNP was performed using Akta Pure system with the Superdex 200 Increase 10/300 GL column (GE Healthcare) for size-exclusion chromatography. Column was equilibrated with 75 ml of HPLC buffer (150 mM NaCl, 30 mM Na3PO4, pH 7.8) at 0.5 ml/min flow rate. The injection volume of a sample was 100 μl, elution volume was 30 ml.
Calibration was done using a series of proteins with known molecular weight (MW). Using the obtained chromatographic data MW for purified recNP A was determined as 57 kDa, that matches the predicted MW of recNP A monomeric form. The chromatogram is shown in Fig. 2.

SDS-PAGE assay
Samples (cell lysates, elution fractions with purified protein, and negative control cell lysates) were mixed with 4x loading buffer containing reducing agent β-mercaptoethanol and incubated at 95°C for 10 min. Then, the mixtures were separated by SDS-PAGE on the 12% separating gel according to the standard procedure and subsequently stained with Coomassie blue R-250.
Western blot analysis
Purified proteins were analyzed by Western blot using antibodies to His tag (Qiagen AntiHis5, #34660) and monoclonal antibodies 4H1 generated to the NP of influenza virus A/Brisbane/10/2007 as well as antibodies 1B12 generated to the NP of B/Brisbane/46/2015 virus obtained in the Smorodintsev Research Institute of Influenza (St. Petersburg, Russia). Proteins were transferred to nitrocellulose membranes using the Bio-Rad Trans Blot Turbo system. Then, the membrane was blocked with non-fat dried milk (NFDM) in PBS buffer containing 0.05% Tween 20 (PBST) for 1 h and incubated with primary antibody solutions (antibodies to His tag or monoclonal antibodies 4H1 and 1B12) in 4% NFDM (Sigma Aldrich) for 2 h following by incubation with secondary antibodies (GAM, Sigma Aldrich, #A3682) for 1 h in PBST buffer.
RESULTS AND DISCUSSION
The NP A and NP B PCR products of influenza A/Brisbane/10/2007 (H3N2) and B/Brisbane/46/2015, respectively, were inserted into the pET22b+ commercial vector (Novagen). The expression vectors pET22b+NP A and pET22b+NP B were designed to produce recNP A and recNP B proteins with a c-terminal 6xHis tag as shown in Fig. 1. The NP A (1496 bp) and NP B (1682 bp) coding sequences were amplified from cDNAs transcribed from the corresponding viral RNAs using the primers shown in the Table 1. As was mentioned hereinabove, the amplification of influenza B NP was more problematic due to a partial homology of the reverse primer to an internal region of the gene (from 299 to 306 bp) leading to the amplification of a shorter product. Therefore, the cloning of the NP of influenza B virus was performed after one PCR round. The sequencing of the plasmids from the corresponding colonies showed the presence of full-length inserts identical to the original sequences.
The pET22b+NP A and pET22b+NP B plasmids were transformed into E. coli BL21(DE3). In the standard induction procedure, cultures were grown for 3 h at 37°C with shaking (250 rpm) in the presence of IPTG (inductor) at a final concentration of 0.1 mM. In order to maximize the yield of soluble NPs, different experimental conditions were screened: various IPTG concentrations (0.05, 0.1, and 0.5 mM) and incubation temperatures (23°C, 32°C, and 37°C). No difference in the protein expression level at different experimental conditions was observed. Therefore, the transformed E. coli were grown at 37°C with the addition of IPTG up to 0.1 mM until the OD600 of culture reached the value of 0.9. Then, the cells were incubated for an additional 3 h. Bacterial cell pellets were obtained by centrifugation at 5,000 g for 15 min (Eppendorf, 5804R), resuspended in lysis buffer, and lysed by sonification using the homogenizer (MSE Soniprep 150, UK).
Supernatants containing soluble fractions of recNPs were purified using Ni-NTA columns and then the products were eluted with high imidazole buffer. Total lysates, fractions, and purified proteins were analyzed by SDSPAGE. This analysis showed the presence of bands of approximately 57 kDa and 62 kDa (Fig. 3) that correlate well with the predicted molecular weight of the recNP fusion proteins of influenza A and B viruses, respectively. After Ni2+ affinity chromatography, the purity of both proteins was above 95%. From 1 liter of the corresponding cultures, 15 mg of NP A and 5 mg of NP B were isolated.

A. NP of influenza A virus: C – negative control, containing E. coli BL21(DE3) with pET22b+, 1total lysate, 2 – soluble fraction, 3 – pellet, 4 – purified recombinant NP, M – molecular marker; B. NP of influenza B virus: C – negative control, containing E. coli BL21(DE3) with pET22b+, 1 – total lysate, 2 – soluble fraction, 3 – pellet, and 4 – purified recombinant NP, M – molecular marker.
Western blot analysis indicated that purified recNP A and recNP B reacted well with the monoclonal antibodies obtained to influenza A/Brisbane/10/2007 and B/Brisbane/46/2015 viruses, respectively. Both proteins also reacted well with anti-His tag antibodies (Qiagen AntiHis5, #34660) (Fig. 4).
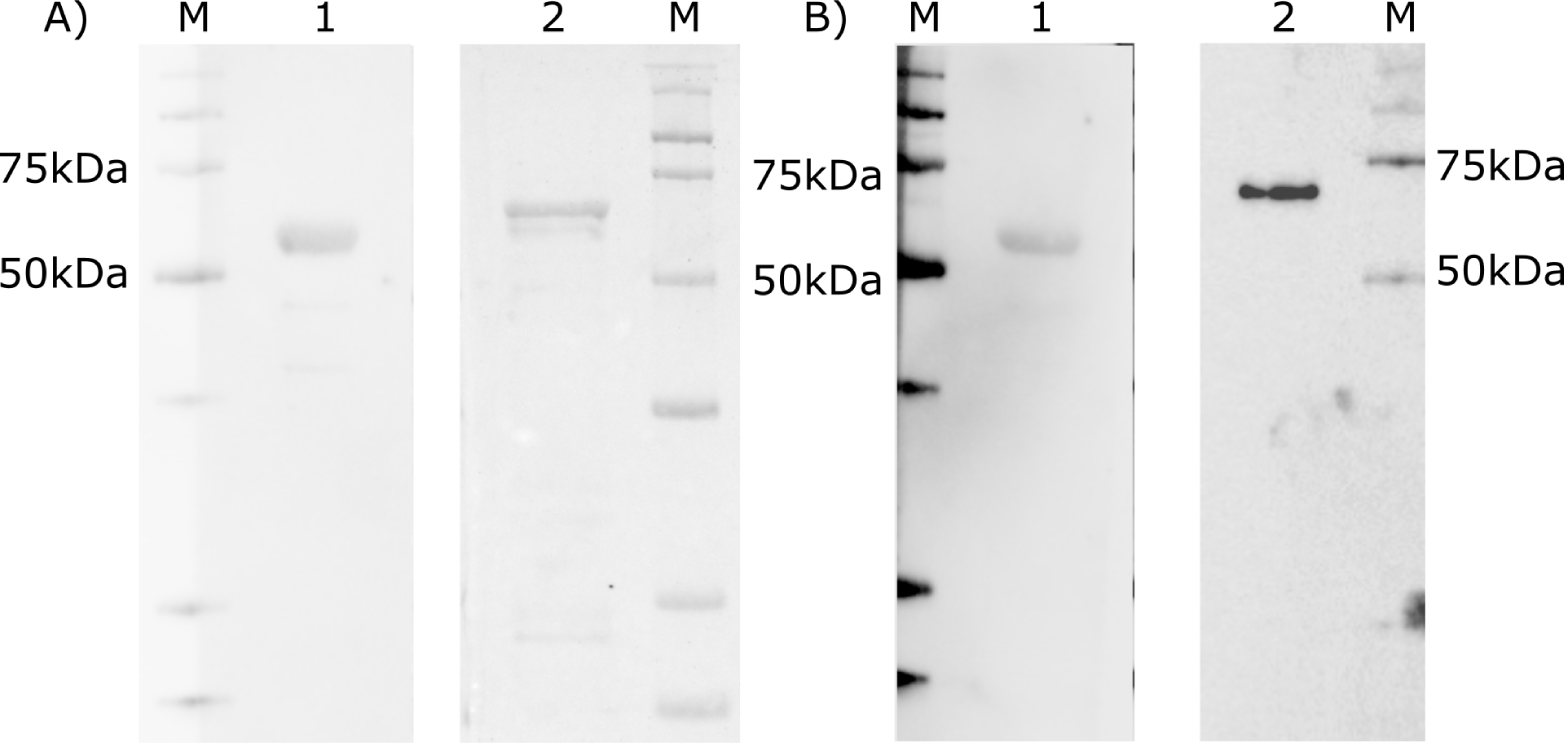
A. Staining with antibodies to 6xHis tag. B. Staining with mouse monoclonal antibodies 4H1 specific to NP of A/Brisbane/10/2007 and 1B12 specific to NP of influenza B/Brisbane/46/2015 virus. M – molecular marker, 1 – NP of A/Brisbane/10/2007, 2 – NP of B/Brisbane/46/2015 virus.
The codon optimization of DNA sequences is widely used in order to increase the expression level of target proteins. A synonymous codon substitutions – substitutions of codons that are translated slower by faster translated codons – can significantly increase the rate of translation of the desired protein [26]. However, it was shown that this procedure can influence the protein folding and stability and can interfere with post-translational protein modification [23, 27]. Some slower translated codons were shown to have advantages for conserving the exact protein structure especially if they encode the amino acids that are located at the key positions of the protein sequence because mutation at these positions would drastically influence the protein properties [28, 29]. Therefore, codon optimization leads not only to the regulation of the protein expression level, but also can influence the structure and function of the expressed proteins. In summary, we showed that influenza A and B heterologous NPs can be successfully expressed in E. coli using original sequences. Both expressed nucleoproteins demonstrated good immune reactivity with specific antibodies.