- Record: found
- Abstract: found
- Article: found
Evolution of In Silico Strategies for Protein-Protein Interaction Drug Discovery
Read this article at
Abstract
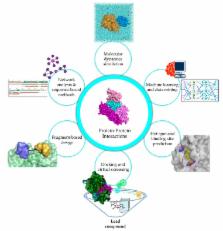
The advent of advanced molecular modeling software, big data analytics, and high-speed processing units has led to the exponential evolution of modern drug discovery and better insights into complex biological processes and disease networks. This has progressively steered current research interests to understanding protein-protein interaction (PPI) systems that are related to a number of relevant diseases, such as cancer, neurological illnesses, metabolic disorders, etc. However, targeting PPIs are challenging due to their “undruggable” binding interfaces. In this review, we focus on the current obstacles that impede PPI drug discovery, and how recent discoveries and advances in in silico approaches can alleviate these barriers to expedite the search for potential leads, as shown in several exemplary studies. We will also discuss about currently available information on PPI compounds and systems, along with their usefulness in molecular modeling. Finally, we conclude by presenting the limits of in silico application in drug discovery and offer a perspective in the field of computer-aided PPI drug discovery.
Related collections
Most cited references318
- Record: found
- Abstract: found
- Article: not found
Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm.
- Record: found
- Abstract: found
- Article: found
Lethality and centrality in protein networks
