- Record: found
- Abstract: found
- Article: found
STIM Protein-NMDA2 Receptor Interaction Decreases NMDA-Dependent Calcium Levels in Cortical Neurons
Read this article at
Abstract
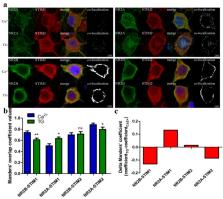
Neuronal Store-Operated Ca 2+ Entry (nSOCE) plays an essential role in refilling endoplasmic reticulum Ca 2+ stores and is critical for Ca 2+-dependent neuronal processes. SOCE sensors, STIM1 and STIM2, can activate Orai, TRP channels and AMPA receptors, and inhibit voltage-gated channels in the plasma membrane. However, the link between STIM, SOCE, and NMDA receptors, another key cellular entry point for Ca 2+ contributing to synaptic plasticity and excitotoxicity, remains unclear. Using Ca 2+ imaging, we demonstrated that thapsigargin-induced nSOCE was inhibited in rat cortical neurons following NMDAR inhibitors. Blocking nSOCE by its inhibitor SKF96365 enhanced NMDA-driven [Ca 2+] i. Modulating STIM protein level through overexpression or shRNA inhibited or activated NMDA-evoked [Ca 2+] i, respectively. Using proximity ligation assays, immunofluorescence, and co-immunoprecipitation methods, we discovered that thapsigargin-dependent effects required interactions between STIMs and the NMDAR2 subunits. Since STIMs modulate NMDAR-mediated Ca 2+ levels, we propose targeting this mechanism as a novel therapeutic strategy against neuropathological conditions that feature NMDA-induced Ca 2+ overload as a diagnostic criterion.
Related collections
Most cited references59
- Record: found
- Abstract: found
- Article: not found
STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx.
- Record: found
- Abstract: found
- Article: not found