- Record: found
- Abstract: found
- Article: not found
Polyglutamine-expanded androgen receptor interferes with TFEB to elicit pathological autophagy defects in SBMA
Read this article at

Abstract
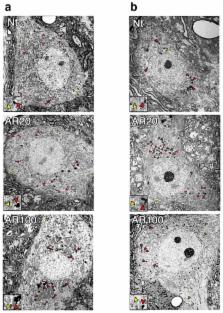
Macroautophagy (hereafter autophagy) is a key pathway in neurodegeneration. Despite protective actions, autophagy may contribute to neuron demise, when dysregulated. Here we considered X-linked spinal and bulbar muscular atrophy (SBMA), a repeat disorder caused by polyglutamine-expanded androgen receptor (polyQ-AR). We found that polyQ-AR reduced long-term protein turnover and impaired autophagic flux in motor neuron-like cells. Ultrastructural analysis of SBMA mice revealed a block in autophagy pathway progression. We considered the transcriptional regulation of autophagy, and observed a functionally significant physical interaction between transcription factor EB (TFEB) and AR. Normal AR promoted, but polyQ-AR interfered with TFEB transactivation. To evaluate physiological relevance, we reprogrammed patient fibroblasts to induced pluripotent stem cells, and then to neuronal precursor cells (NPCs). We compared multiple SBMA NPC lines, and documented metabolic and autophagic flux defects that could be rescued by TFEB. Our results indicate that polyQ-AR diminishes TFEB function to impair autophagy and promote SBMA pathogenesis.
Related collections
Most cited references28
- Record: found
- Abstract: found
- Article: not found
Guidelines for the use and interpretation of assays for monitoring autophagy.
- Record: found
- Abstract: found
- Article: not found
A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells.
- Record: found
- Abstract: found
- Article: not found