- Record: found
- Abstract: found
- Article: found
HIV-1 Vpr activates the G 2 checkpoint through manipulation of the ubiquitin proteasome system
Read this article at
Abstract
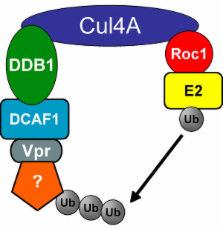
HIV-1 Vpr is a viral accessory protein that activates ATR through the induction of DNA replication stress. ATR activation results in cell cycle arrest in G 2 and induction of apoptosis. In the present study, we investigate the role of the ubiquitin/proteasome system (UPS) in the above activity of Vpr. We report that the general function of the UPS is required for Vpr to induce G 2 checkpoint activation, as incubation of Vpr-expressing cells with proteasome inhibitors abolishes this effect. We further investigated in detail the specific E3 ubiquitin ligase subunits that Vpr manipulates. We found that Vpr binds to the DCAF1 subunit of a cullin 4a/DDB1 E3 ubiquitin ligase. The carboxy-terminal domain Vpr(R80A) mutant, which is able to bind DCAF1, is inactive in checkpoint activation and has dominant-negative character. In contrast, the mutation Q65R, in the leucine-rich domain of Vpr that mediates DCAF1 binding, results in an inactive Vpr devoid of dominant negative behavior. Thus, the interaction of Vpr with DCAF1 is required, but not sufficient, for Vpr to cause G 2 arrest. We propose that Vpr recruits, through its carboxy terminal domain, an unknown cellular factor that is required for G 2-to-M transition. Recruitment of this factor leads to its ubiquitination and degradation, resulting in failure to enter mitosis.
Related collections
Most cited references31
- Record: found
- Abstract: found
- Article: not found
Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery.
- Record: found
- Abstract: found
- Article: not found
ATR regulates fragile site stability.
- Record: found
- Abstract: found
- Article: not found