- Record: found
- Abstract: found
- Article: found
Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation
Read this article at
Abstract
Aims
Mitochondrial dysfunction is a major factor in heart failure (HF). A pronounced variability of mitochondrial electron transport chain (ETC) defects is reported to occur in severe acquired cardiomyopathies without a consistent trend for depressed activity or expression. The aim of this study was to define the defect in the integrative function of cardiac mitochondria in coronary microembolization-induced HF.
Methods and results
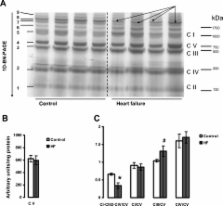
Studies were performed in the canine coronary microembolization-induced HF model of moderate severity. Oxidative phosphorylation was assessed as the integrative function of mitochondria, using a comprehensive variety of substrates in order to investigate mitochondrial membrane transport, dehydrogenase activity and electron-transport coupled to ATP synthesis. The supramolecular organization of the mitochondrial ETC also was investigated by native gel electrophoresis. We found a dramatic decrease in ADP-stimulated respiration that was not relieved by an uncoupler. Moreover, the ADP/O ratio was normal, indicating no defect in the phosphorylation apparatus. The data point to a defect in oxidative phosphorylation within the ETC. However, the individual activities of ETC complexes were normal. The amount of the supercomplex consisting of complex I/complex III dimer/complex IV, the major form of respirasome considered essential for oxidative phosphorylation, was decreased.
Related collections
Most cited references46
- Record: found
- Abstract: found
- Article: not found
Supercomplexes in the respiratory chains of yeast and mammalian mitochondria.
- Record: found
- Abstract: found
- Article: not found
Respiratory complex III is required to maintain complex I in mammalian mitochondria.
- Record: found
- Abstract: found
- Article: not found