
- Record: found
- Abstract: found
- Article: not found
Rationalizing Tight Ligand Binding through Cooperative Interaction Networks
Read this article at

Abstract
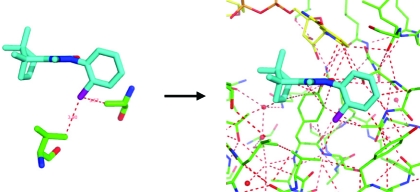
Small modifications of the molecular structure of a ligand sometimes cause strong gains in binding affinity to a protein target, rendering a weakly active chemical series suddenly attractive for further optimization. Our goal in this study is to better rationalize and predict the occurrence of such interaction hot-spots in receptor binding sites. To this end, we introduce two new concepts into the computational description of molecular recognition. First, we take a broader view of noncovalent interactions and describe protein–ligand binding with a comprehensive set of favorable and unfavorable contact types, including for example halogen bonding and orthogonal multipolar interactions. Second, we go beyond the commonly used pairwise additive treatment of atomic interactions and use a small world network approach to describe how interactions are modulated by their environment. This approach allows us to capture local cooperativity effects and considerably improves the performance of a newly derived empirical scoring function, ScorpionScore. More importantly, however, we demonstrate how an intuitive visualization of key intermolecular interactions, interaction networks, and binding hot-spots supports the identification and rationalization of tight ligand binding.
Related collections
Most cited references52
- Record: found
- Abstract: found
- Article: not found
Benchmarking sets for molecular docking.
- Record: found
- Abstract: found
- Article: not found
A critical assessment of docking programs and scoring functions.
- Record: found
- Abstract: found
- Article: not found