- Record: found
- Abstract: found
- Article: found
Testing a MultiTEP-based combination vaccine to reduce Aβ and tau pathology in Tau22/5xFAD bigenic mice
Read this article at
Abstract
Background
Alzheimer disease (AD) is characterized by the accumulation of beta-amyloid (Aβ) plaques and neurofibrillary tangles composed of hyperphosphorylated tau, which together lead to neurodegeneration and cognitive decline. Current therapeutic approaches have primarily aimed to reduce pathological aggregates of either Aβ or tau, yet phase 3 clinical trials of these approaches have thus far failed to delay disease progression in humans. Strong preclinical evidence indicates that these two abnormally aggregated proteins interact synergistically to drive downstream neurodegeneration. Therefore, combinatorial therapies that concurrently target both Aβ and tau might be needed for effective disease modification.
Methods
A combinatorial vaccination approach was designed to concurrently target both Aβ and tau pathologies. Tau22/5xFAD (T5x) bigenic mice that develop both pathological Aβ and tau aggregates were injected intramuscularly with a mixture of two MultiTEP epitope vaccines: AV-1959R and AV-1980R, targeting Aβ and tau, respectively, and formulated in Advax CpG, a potent polysaccharide adjuvant. Antibody responses of vaccinated animals were measured by ELISA, and neuropathological changes were determined in brain homogenates of vaccinated and control mice using ELISA and Meso Scale Discovery (MSD) multiplex assays.
Results
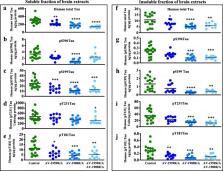
T5x mice immunized with a mixture of Aβ- and tau-targeting vaccines generated high Aβ- and tau-specific antibody titers that recognized senile plaques and neurofibrillary tangles/neuropil threads in human AD brain sections. Production of these antibodies in turn led to significant reductions in the levels of soluble and insoluble total tau, and hyperphosphorylated tau as well as insoluble Aβ 42, within the brains of bigenic T5x mice.
Conclusions
AV-1959R and AV-1980R formulated with Advax CpG adjuvant are immunogenic and therapeutically potent vaccines that in combination can effectively reduce both of the hallmark pathologies of AD in bigenic mice. Taken together, these findings warrant further development of this vaccine technology for ultimate testing in human AD.
Related collections
Most cited references64

- Record: found
- Abstract: found
- Article: found
NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease
- Record: found
- Abstract: found
- Article: not found
Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers.
- Record: found
- Abstract: found
- Article: not found