- Record: found
- Abstract: found
- Article: found
Histone demethylase JMJD1A coordinates acute and chronic adaptation to cold stress via thermogenic phospho-switch
Read this article at
Abstract
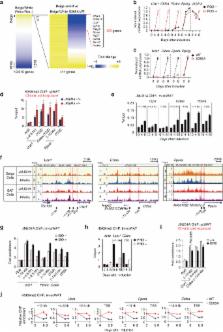
In acute cold stress in mammals, JMJD1A, a histone H3 lysine 9 (H3K9) demethylase, upregulates thermogenic gene expressions through β-adrenergic signaling in brown adipose tissue (BAT). Aside BAT-driven thermogenesis, mammals have another mechanism to cope with long-term cold stress by inducing the browning of the subcutaneous white adipose tissue (scWAT). Here, we show that this occurs through a two-step process that requires both β-adrenergic-dependent phosphorylation of S265 and demethylation of H3K9me2 by JMJD1A. The histone demethylation-independent acute Ucp1 induction in BAT and demethylation-dependent chronic Ucp1 expression in beige scWAT provides complementary molecular mechanisms to ensure an ordered transition between acute and chronic adaptation to cold stress. JMJD1A mediates two major signaling pathways, namely, β-adrenergic receptor and peroxisome proliferator-activated receptor-γ (PPARγ) activation, via PRDM16-PPARγ-P-JMJD1A complex for beige adipogenesis. S265 phosphorylation of JMJD1A, and the following demethylation of H3K9me2 might prove to be a novel molecular target for the treatment of metabolic disorders, via promoting beige adipogenesis.
Abstract
JMJD1A is essential for thermogenic gene induction in brown adipose tissue. Here the authors show that white adipose tissue beige-ing requires both β-adrenergic-dependent phosphorylation of S265 and demethylation activity of JMJD1A while brown adipose tissue-driven thermogenesis requires β-adrenergic dependent phosphorylation of S265 but is independent of H3K9me2 demethylation.
Related collections
Most cited references21
- Record: found
- Abstract: found
- Article: not found
Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Pparγ.
- Record: found
- Abstract: found
- Article: not found
PPARγ agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein.
- Record: found
- Abstract: found
- Article: not found