- Record: found
- Abstract: found
- Article: not found
Copper-Catalyzed Intermolecular Amidation and Imidation of Unactivated Alkanes

Read this article at

Abstract
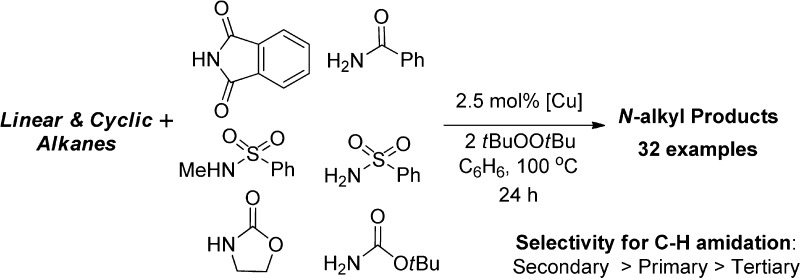
We report a set of rare copper-catalyzed reactions of alkanes with simple amides, sulfonamides, and imides (i.e., benzamides, tosylamides, carbamates, and phthalimide) to form the corresponding N-alkyl products. The reactions lead to functionalization at secondary C–H bonds over tertiary C–H bonds and even occur at primary C–H bonds. [(phen)Cu(phth)] ( 1-phth) and [(phen)Cu(phth) 2] ( 1-phth 2 ), which are potential intermediates in the reaction, have been isolated and fully characterized. The stoichiometric reactions of 1-phth and 1-phth 2 with alkanes, alkyl radicals, and radical probes were investigated to elucidate the mechanism of the amidation. The catalytic and stoichiometric reactions require both copper and tBuOO tBu for the generation of N-alkyl product. Neither 1-phth nor 1-phth 2 reacted with excess cyclohexane at 100 °C without tBuOO tBu. However, the reactions of 1-phth and 1-phth 2 with tBuOO tBu afforded N-cyclohexylphthalimide (Cy-phth), N-methylphthalimide, and tert-butoxycyclohexane (Cy-O tBu) in approximate ratios of 70:20:30, respectively. Reactions with radical traps support the intermediacy of a tert-butoxy radical, which forms an alkyl radical intermediate. The intermediacy of an alkyl radical was evidenced by the catalytic reaction of cyclohexane with benzamide in the presence of CBr 4, which formed exclusively bromocyclohexane. Furthermore, stoichiometric reactions of [(phen)Cu(phth) 2] with tBuOO tBu and (Ph(Me) 2CO) 2 at 100 °C without cyclohexane afforded N-methylphthalimide (Me-phth) from β-Me scission of the alkoxy radicals to form a methyl radical. Separate reactions of cyclohexane and d 12-cyclohexane with benzamide showed that the turnover-limiting step in the catalytic reaction is the C–H cleavage of cyclohexane by a tert-butoxy radical. These mechanistic data imply that the tert-butoxy radical reacts with the C–H bonds of alkanes, and the subsequent alkyl radical combines with 1-phth 2 to form the corresponding N-alkyl imide product.
Related collections
Most cited references30
- Record: found
- Abstract: found
- Article: not found
Cross-dehydrogenative coupling (CDC): exploring C-C bond formations beyond functional group transformations.
- Record: found
- Abstract: not found
- Article: not found
Direct C-H transformation via iron catalysis.
- Record: found
- Abstract: found
- Article: not found