- Record: found
- Abstract: found
- Article: found
α2AP mediated myofibroblast formation and the development of renal fibrosis in unilateral ureteral obstruction
Read this article at
Abstract
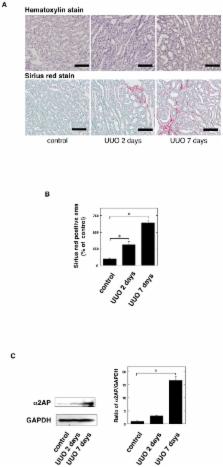
Renal fibrosis is the final common pathway of a wide variety of chronic kidney diseases. Myofibroblast formation via the differentiation of from tissue-resident fibroblasts and bone marrow-derived mesenchymal stem cells (MSCs), and epithelial-to-mesenchymal transition (EMT) is known to play a pivotal role in the development of renal fibrosis. However, the detailed mechanisms underlying this disorder remain unclear. We herein investigated the role of alpha 2-antiplasmin (α2AP) in myofibroblast formation and the development of renal fibrosis. We observed the development of renal fibrosis using unilateral ureteral obstruction (UUO). α2AP had accumulated in the UUO-induced obstructed kidneys and α2AP deficiency attenuated UUO-induced renal fibrosis in mice. The degree of myofibroblast formation in the obstructed kidneys of α2AP −/− mice was less than that in α2AP +/+ mice. In vitro, α2AP induced myofibroblast formation in renal tubular epithelial cells (RTECs), renal fibrosblasts, and bone marrow-derived mesenchymal stem cells (MSCs). α2AP also induced the production of TGF-β, which is known to be a key regulator of myofibroblast formation and fibrosis. α2AP-induced the TGF-β production was significantly reduced by SP600125, c-Jun N-terminal kinase (JNK) specific inhibitor. Our findings suggest that α2AP induces myofibroblast formation in the obstructed kidneys, and mediates the development of renal fibrosis.
Related collections
Most cited references33
- Record: found
- Abstract: found
- Article: not found
Origin and function of myofibroblasts in kidney fibrosis.
- Record: found
- Abstract: found
- Article: not found
Making sense of latent TGFbeta activation.
- Record: found
- Abstract: found
- Article: not found