- Record: found
- Abstract: found
- Article: found
Mitochondrial GSH replenishment as a potential therapeutic approach for Niemann Pick type C disease
Read this article at
Abstract
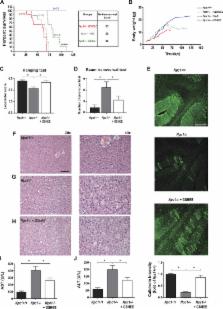
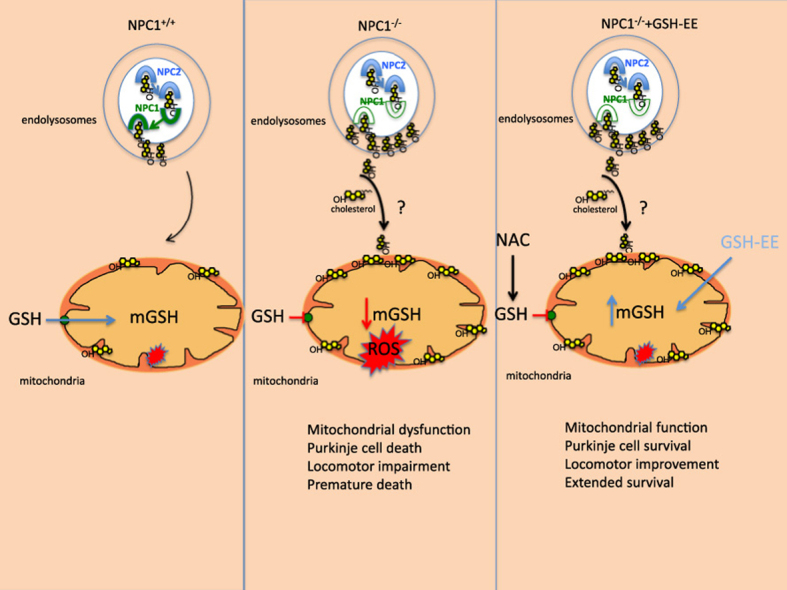
Niemann Pick type C (NPC) disease is a progressive lysosomal storage disorder caused by mutations in genes encoding NPC1/NPC2 proteins, characterized by neurological defects, hepatosplenomegaly and premature death. While the primary biochemical feature of NPC disease is the intracellular accumulation of cholesterol and gangliosides, predominantly in endolysosomes, mitochondrial cholesterol accumulation has also been reported. As accumulation of cholesterol in mitochondria is known to impair the transport of GSH into mitochondria, resulting in mitochondrial GSH (mGSH) depletion, we investigated the impact of mGSH recovery in NPC disease. We show that GSH ethyl ester (GSH-EE), but not N-acetylcysteine (NAC), restored the mGSH pool in liver and brain of Npc1 -/- mice and in fibroblasts from NPC patients, while both GSH-EE and NAC increased total GSH levels. GSH-EE but not NAC increased the median survival and maximal life span of Npc1 -/- mice. Moreover, intraperitoneal therapy with GSH-EE protected against oxidative stress and oxidant-induced cell death, restored calbindin levels in cerebellar Purkinje cells and reversed locomotor impairment in Npc1 -/- mice. High-resolution respirometry analyses revealed that GSH-EE improved oxidative phosphorylation, coupled respiration and maximal electron transfer in cerebellum of Npc1 -/- mice. Lipidomic analyses showed that GSH-EE treatment had not effect in the profile of most sphingolipids in liver and brain, except for some particular species in brain of Npc1 -/- mice. These findings indicate that the specific replenishment of mGSH may be a potential promising therapy for NPC disease, worth exploring alone or in combination with other options.
Graphical abstract
Highlights
-
•
GSH-EE but not NAC restores mitochondrial GSH stores and protects against oxidative stress in NPC disease.
-
•
GSH-EE treatment improves motor coordination and extends survival in Npc1 -/- mice.
-
•
GSH-EE therapy rescues cerebellar mitochondrial dysfunction in Npc1 -/- mice.
-
•
GSH-EE treatment attenuates the liver phenotype in Npc1 -/- mice.
Related collections
Most cited references41

- Record: found
- Abstract: found
- Article: not found
Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis.
- Record: found
- Abstract: found
- Article: not found